Clear Sky Science · es
Variantes de pérdida de función en el activador CAPN1 CD99L2 causan ataxia espástica ligada al cromosoma X
Por qué esto importa para las familias con problemas de movimiento sin explicación
Muchas personas viven años con dificultades para caminar, rigidez muscular o problemas de equilibrio y del habla sin llegar a conocer la causa real. Este estudio muestra cómo las pruebas genéticas modernas pueden, por fin, dar respuestas a algunas de estas familias. Los investigadores no solo compararon diferentes pruebas genéticas para trastornos raros del movimiento, sino que además descubrieron una causa hasta ahora desconocida de una afección llamada ataxia espástica ligada al cromosoma X, lo que apunta a vías biológicas que también pueden ser relevantes en enfermedades cerebrales más comunes.
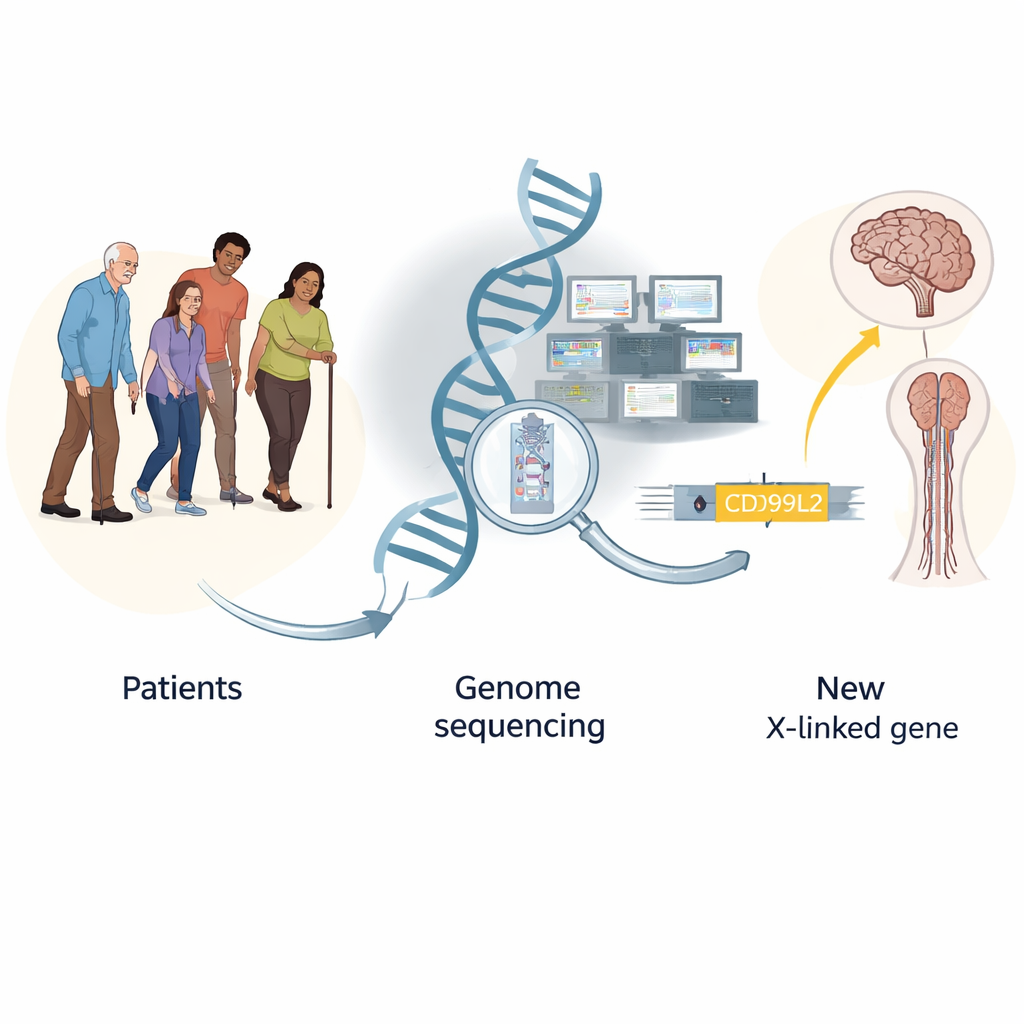
Encontrar las agujas genéticas en un pajar de enfermedades raras
Los trastornos raros del movimiento, como la ataxia (movimientos inestables) y la paraplejía espástica (piernas rígidas y débiles), suelen sospecharse de origen genético, pero en la mayoría de los pacientes las pruebas estándar dan resultado negativo. El equipo siguió a 2.811 personas en Alemania y en toda Europa que fueron remitidas por sospecha de trastornos raros del movimiento durante seis años. Primero analizaron pruebas dirigidas tradicionales que buscan expansiones de repeticiones conocidas en un puñado de genes; estas ofrecieron respuestas en aproximadamente el 11% de los casos. Después emplearon la secuenciación del exoma, que lee solo las partes del genoma que codifican proteínas, y encontraron explicaciones genéticas definitivas en alrededor del 19% de los pacientes, especialmente entre los que presentaban espasticidad.
Mirando más allá de las pruebas estándar con secuenciación del genoma completo
Para avanzar más, los científicos utilizaron la secuenciación del genoma completo, que lee casi todo el ADN de una persona, incluidas regiones que las pruebas estándar y los exomas pueden pasar por alto. Entre 486 individuos que se sometieron a esta prueba más completa, la tasa de diagnóstico aumentó en alrededor de 7,5 puntos porcentuales, en gran parte porque la secuenciación del genoma detecta mejor cambios complejos como reordenamientos estructurales y expansiones de repeticiones. El estudio también mostró que una información clínica bien registrada —especialmente descripciones sintomáticas concretas, edad más joven al realizar la prueba y la combinación de espasticidad con otros problemas del movimiento— ayudó a predecir quiénes tenían más probabilidades de recibir un diagnóstico genético claro.
Descubrir una nueva causa ligada al X de la ataxia espástica
Aun tras estas pruebas extensivas, muchos pacientes permanecieron sin diagnóstico. Los investigadores agruparon datos genéticos de más de 13.000 individuos y utilizaron un enfoque de «carga génica», preguntando qué genes presentaban variantes sospechosas con mayor frecuencia en pacientes que en controles sanos. Este análisis señaló no solo genes de enfermedad ya conocidos, sino que destacó con fuerza un gen hasta entonces poco atendido en el cromosoma X llamado CD99L2. Al combinar resultados de varias familias de Europa, identificaron 25 varones afectados de 20 familias que portaban variantes dañinas en este gen. Estos hombres desarrollaron típicamente problemas para caminar, rigidez en las piernas, habla arrastrada y, a veces, dificultades de equilibrio en la mediana o avanzada edad; las portadoras femeninas permanecieron mayormente sin afectación —patrones que encajan con un trastorno ligado al X. Las variantes destruían principalmente la proteína normal o eliminaban partes cruciales de ella, lo que sugiere con fuerza que la pérdida de su función causa la enfermedad.
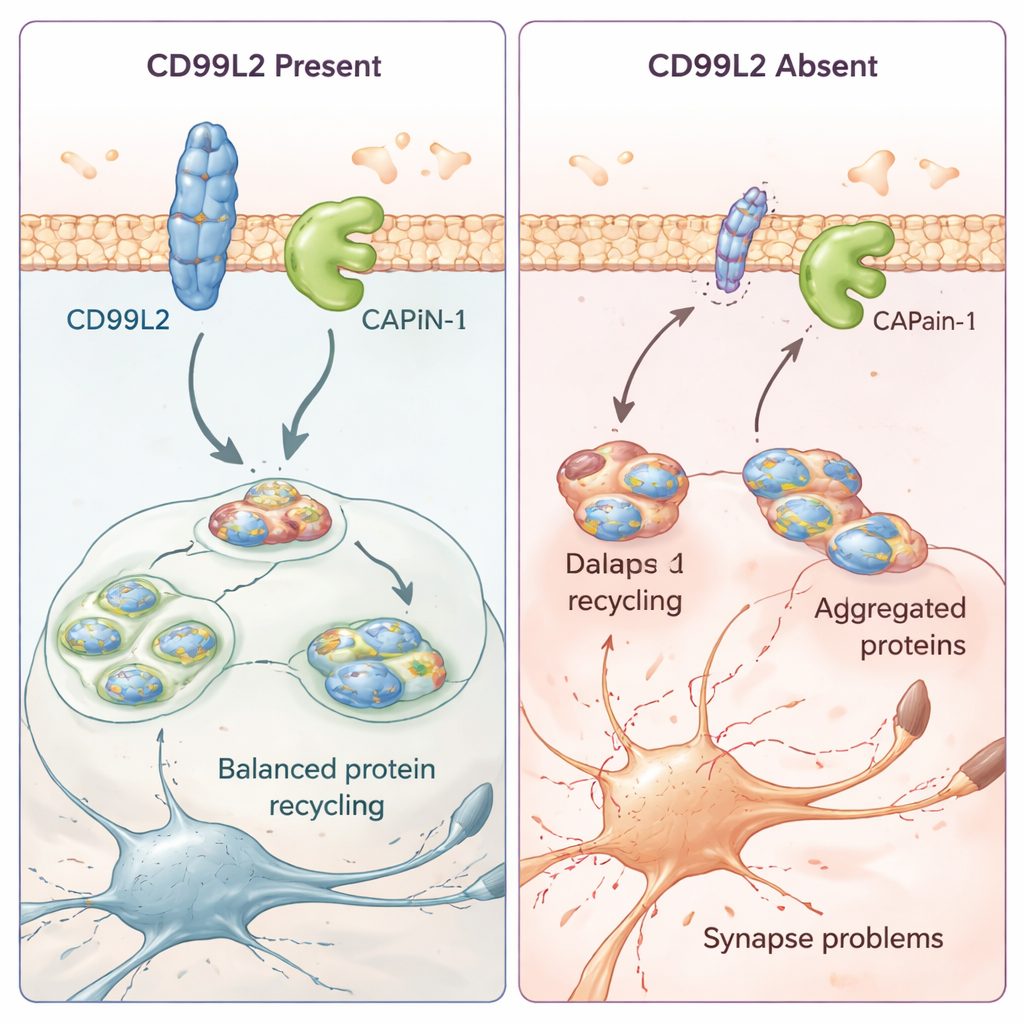
Cómo una pequeña proteína de membrana ayuda a proteger las células cerebrales
Para comprender qué hace realmente CD99L2 en las células, el equipo recurrió a modelos celulares y a células de la piel derivadas de pacientes. Descubrieron que la proteína CD99L2 se localiza en la membrana celular y suele estar marcada con pequeñas etiquetas de «ubiquitina» que controlan cuánto tiempo sobrevive antes de ser degradada. CD99L2 se une físicamente a la calpaína-1 (CAPN1), una enzima activada por calcio que recorta otras proteínas y ayuda a mantener las sinapsis —los puntos de contacto entre neuronas— en buen estado. Cuando CD99L2 está presente e intacta, contribuye a activar y desactivar la calpaína-1 de forma controlada, y luego ella misma es recortada y reciclada. Cuando CD99L2 falta o está alterada estructuralmente, la activación de la calpaína-1 se ve afectada. En las células de los pacientes, esto va acompañado de una actividad alterada de muchos genes relacionados con las sinapsis y la comunicación neuronal, lo que sugiere que cambios sutiles pero generalizados en los circuitos cerebrales pueden subyacer a los problemas de movimiento progresivos.
Qué significa esto para los pacientes hoy y mañana
Para las familias con ataxia espástica o paraplejía espástica sin explicación, este trabajo ofrece dos tipos de progreso. Primero, demuestra que usar la secuenciación del genoma completo de forma temprana, junto con una descripción clínica cuidadosa, puede aumentar notablemente las posibilidades de obtener un diagnóstico genético firme. Segundo, añade a CD99L2 a la lista de genes que regulan la actividad de las calpaínas, una vía ya implicada en otras ataxias raras y en enfermedades comunes como el Alzheimer y el Parkinson. En términos cotidianos, el estudio revela un nuevo interruptor de ‘‘encendido-apagado’’ que ayuda a mantener el equilibrio del mantenimiento celular en el cerebro; cuando ese interruptor se rompe, las neuronas se deterioran lentamente, dando lugar a rigidez y mala coordinación. Comprender este interruptor podría, en el futuro, abrir la puerta a tratamientos que ajusten la actividad de las calpaínas y protejan las células cerebrales en un rango de enfermedades neurológicas.
Cita: Menden, B., Incebacak Eltemur, R.D., Demidov, G. et al. Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia. Nat Commun 17, 1698 (2026). https://doi.org/10.1038/s41467-026-69337-9
Palabras clave: ataxia espástica, trastornos del movimiento raros, secuenciación del genoma, CD99L2, calpaína-1