Clear Sky Science · es
Conjuntos a resolución atómica de proteínas intrínsecamente desordenadas con AlphaFold
Por qué importan las proteínas camaleónicas
Nuestras células están llenas de proteínas que nunca se asientan en una única forma rígida. Estas proteínas “intrínsecamente desordenadas” se comportan más como fideos flexibles que como máquinas plegadas con precisión, y sin embargo son centrales en procesos que van desde la señalización celular hasta las enfermedades neurodegenerativas. Debido a que se mueven y flexionan constantemente, capturar todo su abanico de conformaciones a nivel atómico es extremadamente difícil y suele requerir años de experimentos y cálculos intensivos. Este artículo presenta una nueva forma de aprovechar la inteligencia artificial y la física juntas para mapear estas moléculas inquietas de manera mucho más eficiente.
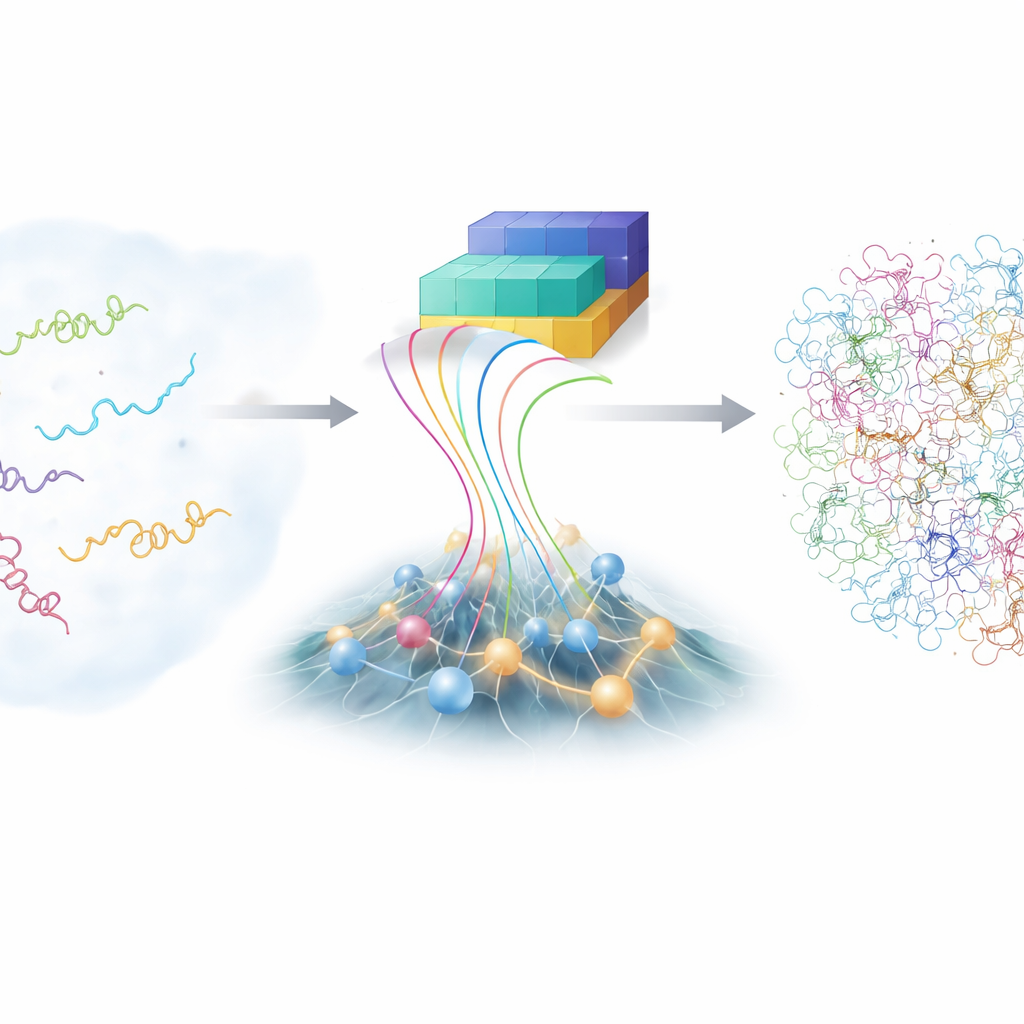
El reto de las moléculas inquietas
A diferencia de los modelos de libro de texto que muestran una estructura ordenada, las proteínas intrínsecamente desordenadas (PID) recorren un vasto paisaje de formas posibles. Esa flexibilidad les ayuda a reconocer muchos socios distintos, pero también las hace notoriamente difíciles de estudiar. Las técnicas de laboratorio tradicionales, como la resonancia magnética nuclear avanzada y la dispersión de rayos X, pueden informar sobre promedios de muchas conformaciones, pero no sobre cada forma individual. Las simulaciones informáticas con detalle atómico pueden, en principio, seguir cada átomo mientras una PID se retuerce, pero son extremadamente costosas y dependen de modelos físicos finamente ajustados. Como resultado, la comunidad científica dispone solo de una colección limitada de conjuntos precisos y detallados de PID para aprender.
Combinar conjeturas inteligentes con reglas físicas
En los últimos años, la familia de herramientas de aprendizaje profundo AlphaFold ha sorprendido a la biología al predecir estructuras de proteínas a partir de sus secuencias de aminoácidos. Para las proteínas desordenadas, sin embargo, la fortaleza habitual de AlphaFold —adivinar una única mejor forma— es menos útil, porque las PID no tienen solo una. Lo que AlphaFold sí proporciona es información rica sobre la probabilidad de que distintas partes de la cadena estén cerca o lejos entre sí. Los autores construyeron un nuevo marco, llamado bAIes, que trata esta información derivada de la IA como una guía flexible y la mezcla con un modelo físico rápido que parte deliberadamente de una visión de “coil aleatorio”, donde la cadena explora todos los pliegues y giros posibles sin favorecer ninguna estructura concreta.
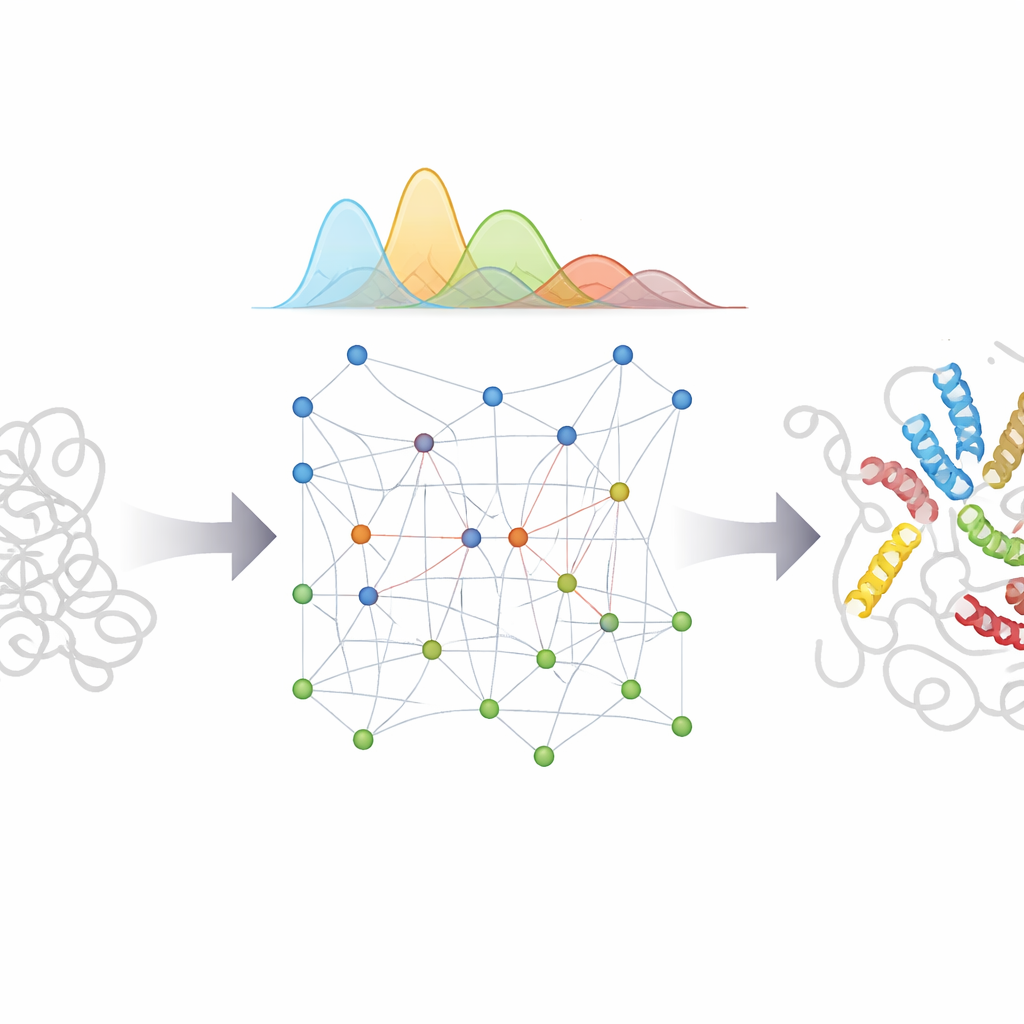
De enredos aleatorios a conjuntos realistas
Primero, los investigadores construyeron un modelo físico eficiente que reproduce cómo se comporta una cadena proteica completamente no estructurada, basándose en estadísticas extraídas de miles de estructuras proteicas conocidas. Este modelo sirve como el “prior” —la expectativa básica de cómo se mueve una PID si no sabemos nada más. A continuación, bAIes interpreta las predicciones de AlphaFold sobre qué pares de residuos tienden a acercarse. En lugar de forzar a la proteína a adoptar un único patrón, convierte esas pistas en restricciones de distancia suaves con incertidumbre incorporada, permitiendo que la cadena satisfaga las sugerencias de la IA solo cuando son consistentes con el panorama físico más amplio.
Probar frente a experimentos reales
Para evaluar si este enfoque funciona, el equipo aplicó bAIes a un conjunto de 21 proteínas que iban desde coils casi completamente aleatorios hasta sistemas más complejos con hélices transitorias y múltiples dominios. Para cada una, compararon los conjuntos generados por ordenador con una amplia gama de mediciones experimentales que sondan tanto detalles locales como el tamaño y la forma globales. Para proteínas muy flexibles, como el péptido relacionado con el Alzheimer Aβ40, el simple modelo de coil aleatorio ya estaba bastante cerca de la realidad, y bAIes preservó ese buen acuerdo. Para proteínas parcialmente estructuradas, bAIes mejoró la concordancia con los experimentos al captar correctamente dónde aparecen y desaparecen segmentos helicoidales cortos y parches compactos. Es crucial que el método se mantuviera robusto incluso cuando AlphaFold mostraba exceso de confianza y predecía erróneamente pliegues estables donde los experimentos en solución muestran desorden, porque bAIes permite explícitamente errores en la entrada de la IA.
Superando o igualando métodos existentes
Los autores compararon entonces bAIes con simulaciones atómicas completas ejecutadas en superordenadores especializados, con modelos mesoscópicos principales que simplifican las proteínas a cuentas, y con nuevos generadores de aprendizaje profundo entrenados con datos de simulación. En múltiples pruebas, bAIes igualó o superó de forma consistente a estos enfoques en la reproducción de datos experimentales, al tiempo que requería mucha menos potencia computacional que las simulaciones a gran escala. También funcionó más allá de las PID simples, manejando proteínas con varios dominios rígidos unidos por conectores flexibles y recuperando sus formas globales en solución. Cuando los investigadores afinaron adicionalmente los conjuntos de bAIes con datos experimentales, el acuerdo mejoró aún más, mostrando que el método puede servir como un punto de partida potente para el modelado integrador.
Qué significa esto para la biología y la medicina
Al casar el poder de reconocimiento de patrones de AlphaFold con un modelo físico cuidadosamente diseñado y un tratamiento bayesiano de la incertidumbre, bAIes ofrece una vía práctica hacia “películas” detalladas de proteínas desordenadas en lugar de instantáneas únicas. Estos conjuntos con detalle atómico pueden ayudar a los científicos a entender cómo las regiones flexibles reconocen a sus socios, cómo comienzan el mal plegamiento y la agregación en enfermedades como el Parkinson y el Alzheimer, y cómo pequeñas moléculas podrían unirse a objetivos esquivos y cambiantes. Dado que el método es eficiente y está implementado en software de código abierto, puede adoptarse ampliamente para generar conjuntos realistas de muchas proteínas desordenadas, guiando experimentos y apoyando futuros sistemas de IA que aspiren a predecir no solo una estructura, sino el rango completo de formas que las moléculas más flexibles de la vida pueden adoptar.
Cita: Schnapka, V., Morozova, T.I., Sen, S. et al. Atomic resolution ensembles of intrinsically disordered proteins with Alphafold. Nat Commun 17, 2399 (2026). https://doi.org/10.1038/s41467-026-69172-y
Palabras clave: proteínas intrínsecamente desordenadas, AlphaFold, modelado bayesiano, conjuntos de proteínas, biología estructural