Clear Sky Science · es
scLong: un modelo fundacional de miles de millones de parámetros para capturar el contexto génico a largo alcance en transcriptómica unicelular
Enseñar a las computadoras a leer el lenguaje oculto de las células
Cada célula de tu cuerpo alberga una ciudad en plena actividad donde los genes se encienden y apagan siguiendo patrones intrincados. La secuenciación de ARN unicelular moderna puede ahora escuchar cada célula individual, pero el resultado es una avalancha abrumadora de números. Este artículo presenta scLong, un enorme modelo de inteligencia artificial diseñado para dar sentido a estos complejos patrones de actividad génica, incluidas las señales débiles que los métodos anteriores tienden a ignorar. Su objetivo es ayudar a los investigadores a entender cómo reaccionan las células cuando se alteran genes, se administran fármacos o se desarrolla una enfermedad.
Por qué importan los mapas génicos a nivel celular
Los estudios génicos tradicionales suelen mezclar millones de células, promediando las raras o inusuales. Las técnicas unicelulares cambiaron eso al medir la actividad génica en cada célula por separado, revelando tipos celulares ocultos, comunicaciones sutiles entre células y circuitos de control detallados que deciden la función celular. Sin embargo, analizar este tipo de datos es extremadamente desafiante: cada célula puede tener niveles de actividad medidos para decenas de miles de genes, muchos apenas detectables. Los modelos de IA existentes simplifican el problema centrándose solo en los genes más ruidosos, lo que acelera el cálculo pero pierde muchas señales sutiles que podrían ser cruciales en enfermedad, desarrollo o respuesta a fármacos.
Un nuevo modelo que escucha a todos los genes
scLong afronta este reto ampliando en lugar de recortar. Es un modelo fundacional de miles de millones de parámetros entrenado con perfiles de actividad génica de alrededor de 48 millones de células humanas procedentes de más de 50 tejidos. A diferencia de enfoques anteriores que atienden a unos pocos miles de genes muy activos, scLong considera aproximadamente 28.000 genes a la vez, incluidos aquellos que se expresan rara o débilmente. Combina dos tipos de información para cada gen: su nivel de actividad en una célula dada y lo que ya se sabe sobre su función a partir de la Gene Ontology, un amplio catálogo curado por expertos sobre roles y relaciones génicas. Una red especializada que opera sobre un grafo de conexiones génicas destila este conocimiento previo en representaciones compactas que el modelo puede usar junto con los valores de expresión bruta.
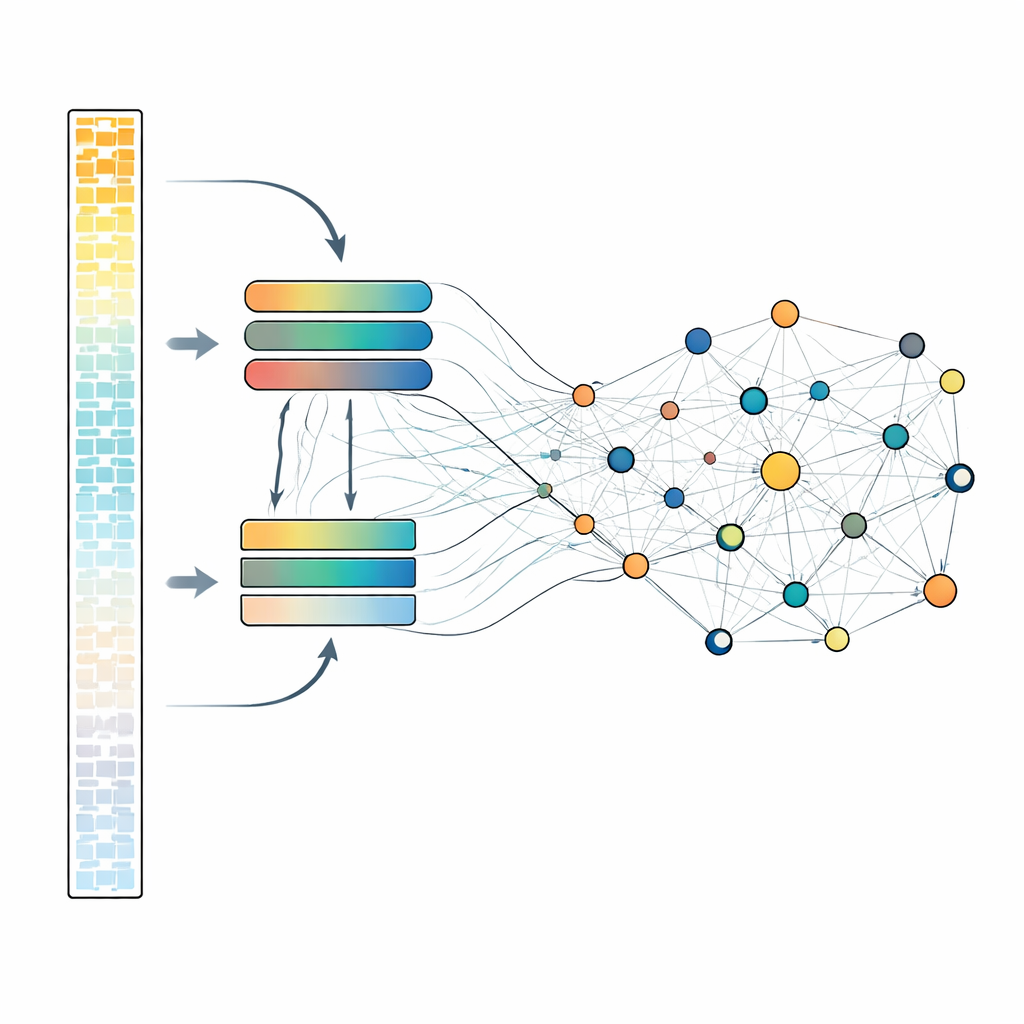
Cómo el modelo equilibra potencia y eficiencia
Examinar cada gen en detalle es costoso computacionalmente, por lo que scLong emplea un diseño inteligente de dos vías. Dentro de cada célula, los genes se ordenan según la intensidad de su expresión. Los genes más activos, que a menudo contienen la señal biológica principal, se procesan mediante un módulo de atención mayor y más potente. Los genes más silenciosos, incluyendo mediciones bajas o incluso cero, pasan por un módulo más pequeño y ligero. Después, todos los genes se reunen y atraviesan otra capa de atención que permite que cada gen influya en todos los demás. Este diseño permite al modelo mantener representaciones más baratas pero aún significativas para las señales débiles mientras reserva más capacidad para las más fuertes. Durante el preentrenamiento, el sistema oculta repetidamente un subconjunto de valores de actividad génica y aprende a reconstruirlos a partir del contexto circundante, obligándolo a descubrir los patrones que vinculan a los genes.
Aplicando el modelo a problemas reales
Una vez entrenado, scLong puede adaptarse a una amplia gama de preguntas biológicas. Los autores muestran que predice cómo cambiará la actividad génica cuando genes específicos se desactiven o muten, incluidas combinaciones de dos genes que pueden actuar conjuntamente. También pronostica cómo responden las células cuando se exponen a distintos compuestos químicos, lo cual es importante para el descubrimiento de fármacos y las pruebas de seguridad. En estudios de cáncer, scLong ayuda a anticipar cómo líneas celulares tumorales responderán a fármacos individuales y a pares de fármacos que podrían funcionar mejor en combinación, superando con frecuencia tanto a modelos especializados como a otros grandes modelos fundacionales. Más allá de la predicción, scLong puede inferir redes de relaciones regulatorias entre genes y contribuir a corregir distorsiones técnicas que surgen cuando los datos se recogen en distintos laboratorios o con distintas máquinas.
Qué implica esto para la medicina y la investigación futuras
En términos sencillos, scLong ofrece a los científicos un mapa de alta resolución y consciente del contexto de la actividad génica dentro de células individuales, uno que no descarta los genes silenciosos o poco utilizados. Al aprender de millones de células e incorporar conocimiento biológico existente, proporciona estimaciones más precisas sobre cómo reaccionarán las células cuando se alteren genes, se introduzcan nuevos fármacos o avancen procesos patológicos. Esto podría acelerar la búsqueda de nuevas terapias, orientar decisiones de tratamiento más personalizadas y afinar nuestra comprensión de cómo redes génicas complejas controlan la salud y la enfermedad. Aunque el modelo es grande y exige recursos computacionales, apunta hacia un futuro en el que sistemas de IA potentes y de propósito general sirvan como compañeros versátiles para explorar los mecanismos ocultos de nuestras células.
Cita: Bai, D., Mo, S., Zhang, R. et al. scLong: a billion-parameter foundation model for capturing long-range gene context in single-cell transcriptomics. Nat Commun 17, 2380 (2026). https://doi.org/10.1038/s41467-026-69102-y
Palabras clave: transcriptómica unicelular, modelos fundacionales, regulación génica, predicción de respuesta a fármacos, expresión génica