Clear Sky Science · es
Descubriendo la diversidad de PAM de Cas9 mediante minería metagenómica y aprendizaje automático
Por qué importa esto para la edición génica futura
CRISPR se ha convertido en un símbolo de la edición génica moderna, pero una regla silenciosa sigue limitando lo que puede hacer: cada corte en el ADN debe estar junto a una corta secuencia “permiso” . Estas secuencias cortas, llamadas PAM, deciden dónde la popular enzima Cas9 puede y no puede actuar. Este estudio muestra cómo rastrear enormes cantidades de ADN microbiano, combinado con aprendizaje automático avanzado, puede revelar una enorme variedad oculta de estos permisos. Ese nuevo mapa podría abrir muchos más lugares en el genoma humano a terapias precisas y más seguras.
Reglas ocultas que guían los cortes de CRISPR
Cas9 y enzimas relacionadas forman parte de un sistema inmune natural presente en bacterias y arqueas. Para evitar cortar su propio ADN, estos microbios hacen que las proteínas Cas busquen un PAM—un tramo muy corto de letras—junto al sitio objetivo. Solo cuando ese PAM está presente, Cas9 desenrolla el ADN y permite que su ARN guía compruebe si hay coincidencia, provocando un corte si todo encaja. El problema para la medicina es que los caballos de laboratorio comunes, como la Cas9 estándar de Streptococcus pyogenes, reconocen solo patrones de PAM estrechos. Si una mutación causante de enfermedad carece de la secuencia adecuada cercana, las herramientas actuales simplemente no pueden alcanzarla sin sacrificar precisión.
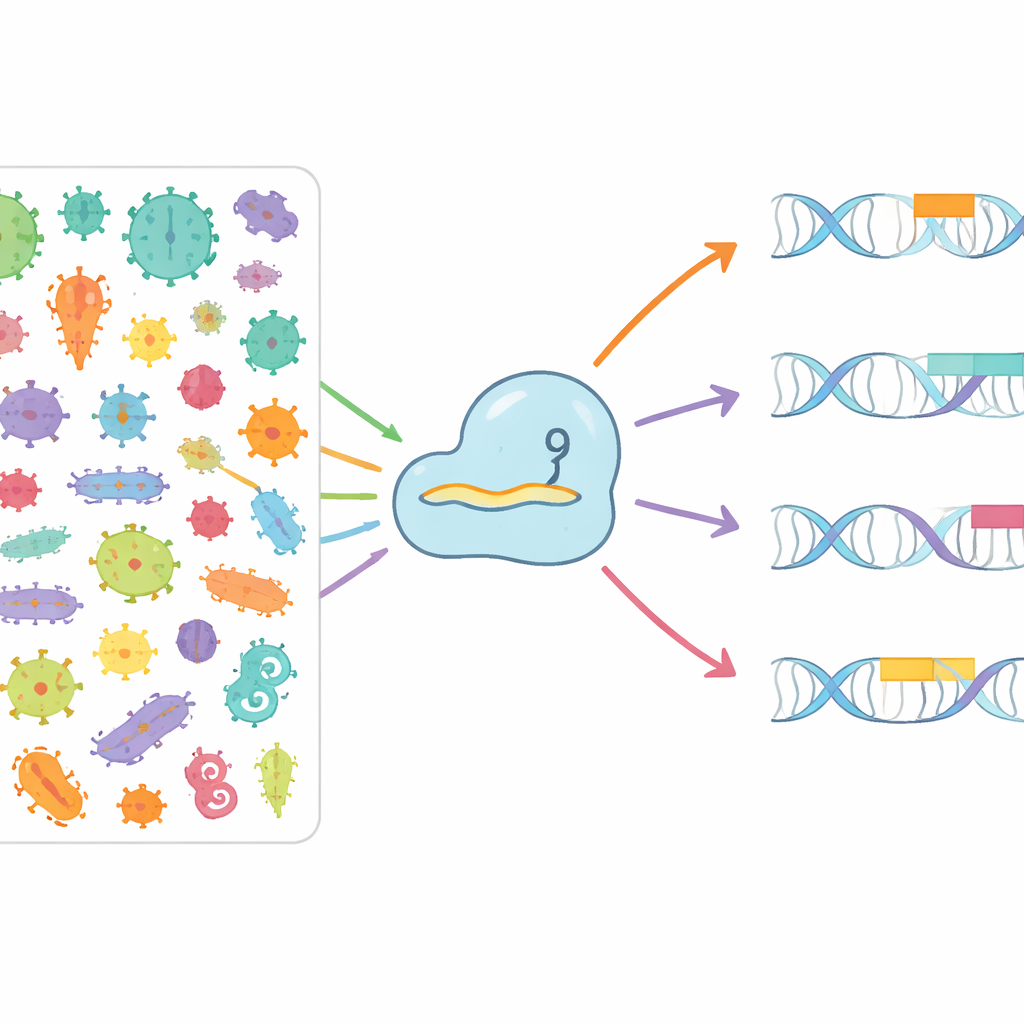
Minería del mundo microbiano en busca de nuevas opciones
Los autores se propusieron cartografiar sistemáticamente cómo diferentes proteínas Cas9 reconocen distintos PAM en la naturaleza. Revisaron más de 3,8 millones de genomas bacterianos y arqueales, y más de 7,4 millones de secuencias virales y de plásmidos que infectan o se mueven entre microbios. Al identificar arreglos CRISPR, vincularlos a genes Cas9 cercanos y luego emparejar los espaciadores “de memoria” almacenados contra virus y plásmidos invasores, pudieron ver qué patrones cortos de ADN tendían a flanquear objetivos reales. A partir de esto construyeron CRISPR-PAMdb, un catálogo público que contiene 8003 grupos de Cas9, cada uno emparejado con un perfil consensuado de PAM, y los organizaron en un árbol evolutivo que destaca cómo las enzimas Cas9 relacionadas tienden a compartir preferencias de PAM similares, aunque mostrando una diversidad general notable.
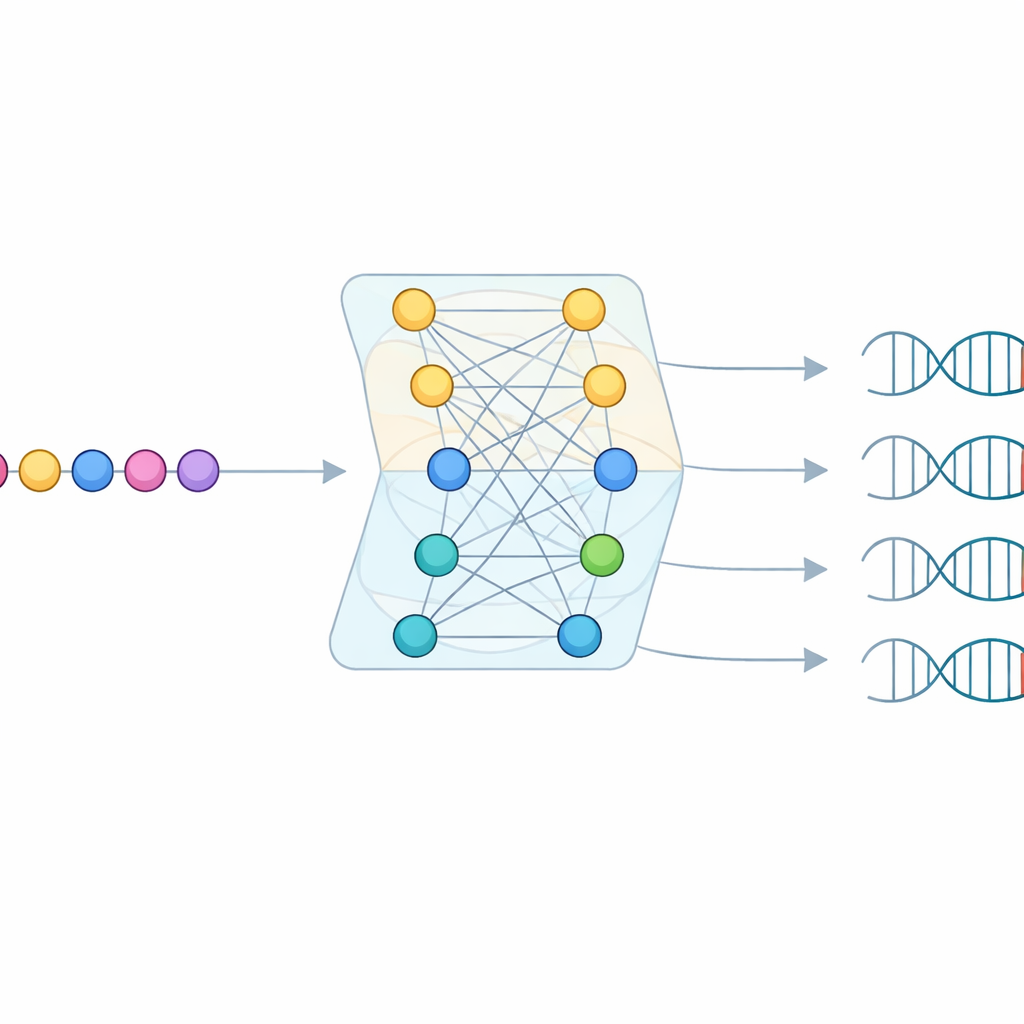
Cuando los datos se agotan, deja que el modelo aprenda
Incluso con este sondeo gigantesco, la mayoría de las proteínas Cas9 que encontraron carecían de suficientes objetivos virales coincidentes para leer un PAM directamente. Para cubrir esos huecos, el equipo construyó un modelo de aprendizaje automático llamado CICERO. CICERO usa un potente “modelo de lenguaje” de proteínas que ha aprendido patrones generales de secuencias de aminoácidos y lo afina para predecir, para cualquier proteína Cas9 dada, cuán probable es que cada letra del ADN aparezca en cada una de diez posiciones del PAM. El modelo se entrenó con los perfiles de PAM de CRISPR-PAMdb y luego se probó tanto por validación cruzada como en 79 enzimas Cas9 cuyas PAM se habían medido experimentalmente, logrando una fuerte concordancia entre predicción y realidad.
Saber cuán confiables son las predicciones
Una característica clave de CICERO es que no se limita a adivinar un PAM: también estima cuán fiable es cada conjetura. Tras aprender a predecir patrones de PAM, los investigadores entrenaron una segunda red ligera que toma la misma secuencia de Cas9 y aprende a pronosticar cuán precisa será la predicción del PAM. Las puntuaciones de confianza más altas se correspondieron fuertemente con una mayor precisión en el mundo real. Usando este filtro de confianza, el equipo amplió las anotaciones de PAM a más de 50.000 proteínas Cas9 adicionales, con más de 17.000 predicciones clasificadas como de alta confianza. Esto amplía enormemente el catálogo de variantes Cas9 con reglas de direccionamiento razonablemente bien comprendidas.
Qué significa esto para tratar enfermedades genéticas
Para mostrar por qué importan estos nuevos recursos, los autores examinaron decenas de miles de mutaciones de una sola letra vinculadas a enfermedades en la base de datos ClinVar que, en principio, podrían corregirse usando editores de bases—herramientas que cambian una letra del ADN sin romper ambas hebras. Encontraron que la enzima Cas9 estándar solo puede acceder a alrededor de la mitad de esos sitios debido a sus estrictas exigencias de PAM. Cuando permitieron el uso de parientes de Cas9 de CRISPR-PAMdb y predicciones de CICERO de alta confianza que reconocen un conjunto más amplio pero aún específico de secuencias cercanas, casi todas estas mutaciones se volvieron teóricamente accesibles sin relajar el direccionamiento hasta el punto de perder precisión.
Una caja de herramientas mayor para la cirugía precisa del ADN
En términos sencillos, este trabajo construye dos cosas: un mapa público gigante que vincula miles de proteínas Cas9 naturales con los cortos patrones de ADN que prefieren, y una guía de IA que puede predecir esas preferencias para muchas más enzimas solo a partir de sus secuencias. Juntos, convierten el mundo microbiano en una rica biblioteca de piezas para futuros editores genéticos. A medida que los investigadores refinen y prueben estas variantes Cas9 en el laboratorio, los clínicos podrían disponer de herramientas más seguras y versátiles que alcancen mutaciones causantes de enfermedad que antes estaban fuera de alcance, acercando la cirugía genómica verdaderamente precisa un paso más a la realidad.
Cita: Fang, T., Bogensperger, L., Feer, L. et al. Uncovering Cas9 PAM diversity through metagenomic mining and machine learning. Nat Commun 17, 2510 (2026). https://doi.org/10.1038/s41467-026-69098-5
Palabras clave: CRISPR-Cas9, diversidad de PAM, metagenómica, aprendizaje automático, edición del genoma