Clear Sky Science · es
Predicción de similitud química entre espectros de masas en modos de ionización cruzada en metabolómica
Por qué importa conectar los puntos químicos
Cada sorbo de café, bocanada de aire o dosis de medicamento deja trazas químicas en nuestro cuerpo. Los instrumentos modernos pueden detectar miles de estas moléculas a la vez, pero convertir esas señales en conocimiento biológico sigue siendo sorprendentemente difícil. Este estudio presenta MS2DeepScore 2.0, una herramienta de aprendizaje automático que ayuda a los científicos a ver cómo se relacionan estas moléculas, incluso cuando las señales se registran de formas muy diferentes. Al hacerlo, promete interpretaciones más rápidas y completas de mezclas químicas complejas en medicina, nutrición e investigación ambiental.
Dos formas de mirar la misma molécula
La espectrometría de masas es una técnica fundamental que pesa y fragmenta moléculas para revelar su identidad. En los experimentos rutinarios, los científicos suelen medir la misma muestra en dos modos: uno que favorece moléculas cargadas positivamente y otro que favorece las cargadas negativamente. Cada modo produce su propio “código de barras” característico de fragmentos. Incluso cuando ambas mediciones provienen de la misma molécula, los patrones resultantes pueden ser tan diferentes que los métodos de comparación tradicionales fallan. Como resultado, los investigadores suelen analizar los dos modos por separado, construir dos mapas desconectados de la muestra y correr el riesgo de perder relaciones químicas importantes.
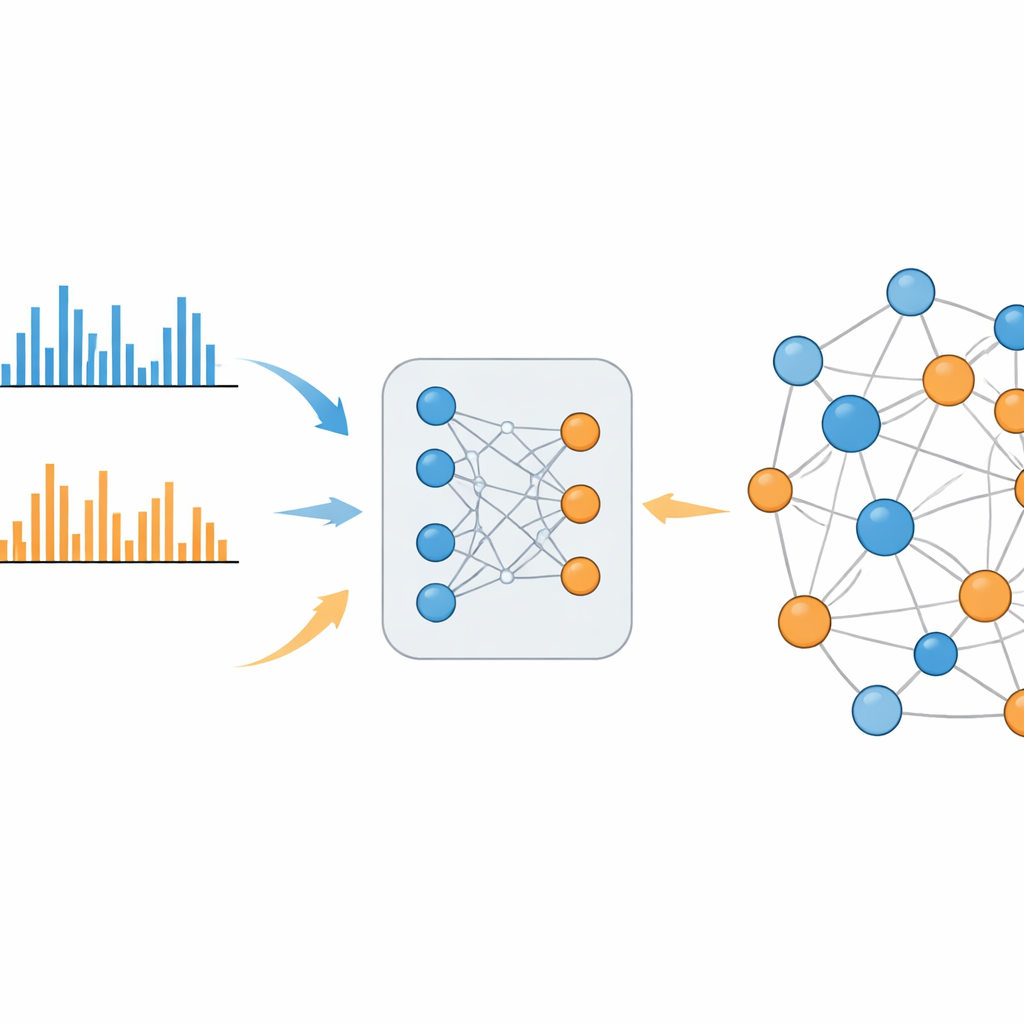
Un sistema que salva la brecha
MS2DeepScore 2.0 aborda esta división aprendiendo la similitud química directamente de grandes bibliotecas de espectros conocidos. El modelo se basa en un diseño de red neuronal gemela que convierte cada patrón de fragmentación en una huella de 500 números, llamada embedding. Durante el entrenamiento, el sistema ve cientos de miles de ejemplos de modos positivos y negativos, junto con la similitud real entre las moléculas subyacentes. Se ajusta de modo que los espectros de moléculas relacionadas terminen con embeddings similares, ya sea que se midieran en el mismo modo o en modos opuestos. La nueva versión va más allá de su predecesora al incorporar información adicional, como la masa de la molécula original y el modo de ionización utilizado, y al emplear un esquema de muestreo cuidadosamente equilibrado para que las relaciones químicas raras pero informativas no queden ahogadas por las comunes y poco informativas.
De señales dispersas a mapas unificados
Una vez entrenado, MS2DeepScore 2.0 puede estimar cuán químicamente similares son dos espectros, incluidos pares positivo versus negativo. Los autores muestran que estas predicciones se correlacionan bien con medidas establecidas de similitud estructural, no solo dentro de cada modo sino también entre modos. Usando datos reales de orina humana, plasma sanguíneo humano y una planta comestible silvestre, construyen “redes moleculares” en las que cada espectro es un nodo y una fuerte similitud predicha crea una conexión. A diferencia de enfoques anteriores, estas redes mezclan de forma natural datos de modos positivos y negativos en mapas únicos y coherentes. Clústeres curados por expertos incluyen, por ejemplo, grupos de moléculas relacionadas con la cafeína en orina que están conectados a través de modos de ionización y coinciden con vías metabólicas conocidas.
Ver el paisaje químico de un vistazo
Las redes moleculares son potentes pero pueden enredarse si se incluyen demasiados enlaces débiles. Para evitar esto, los autores usan los embeddings de MS2DeepScore directamente como coordenadas en una disposición bidimensional creada con una técnica llamada UMAP. Cada punto en este mapa representa un espectro, y los puntos cercanos corresponden a moléculas que el modelo considera químicamente similares. Los espectros de modo positivo y negativo de un mismo compuesto, que a simple vista se ven muy diferentes, a menudo acaban lado a lado en este espacio de embeddings. El equipo también entrena un modelo adicional que inspecciona cada embedding y estima cuán fiable es, señalando espectros que son ruidosos, incompletos o distintos a todo lo visto durante el entrenamiento. Eliminar estos puntos de baja calidad mejora la precisión general y hace que las visualizaciones sean más fiables.
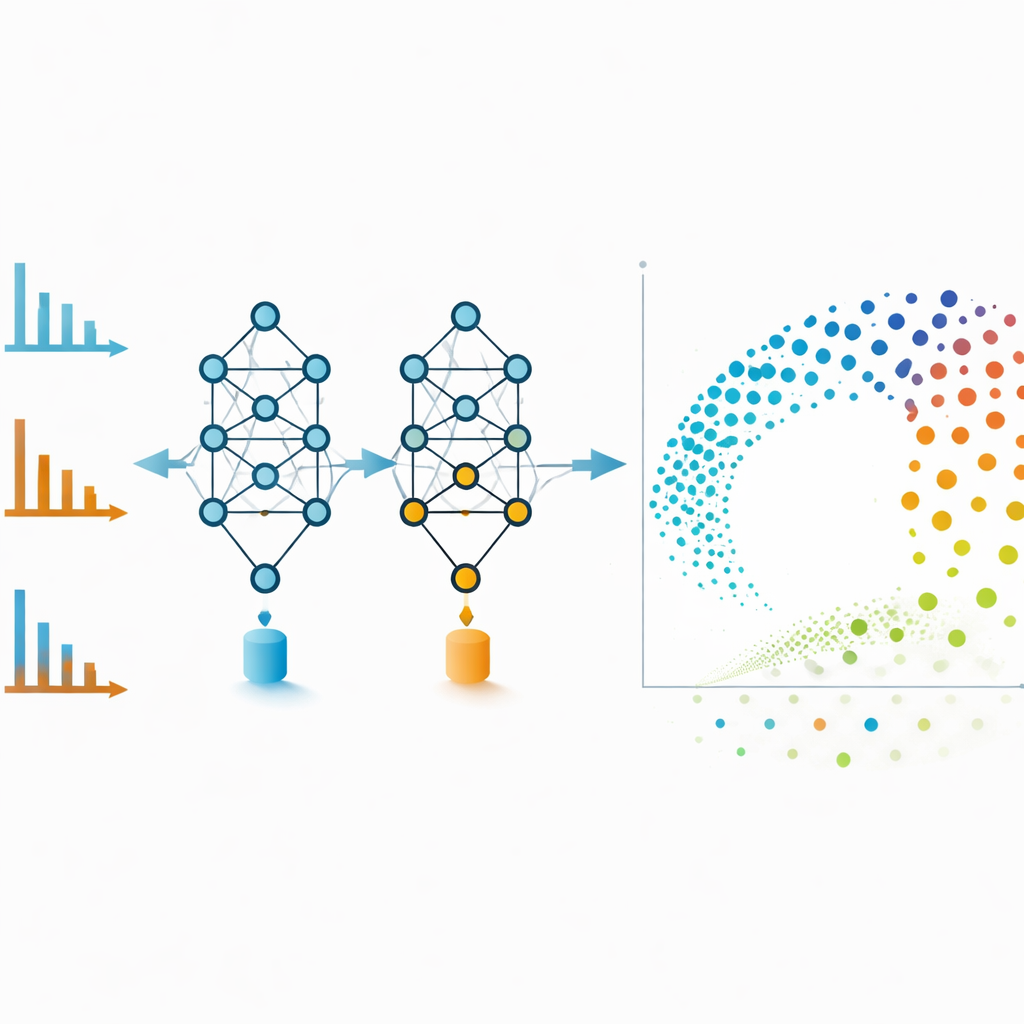
Llevar herramientas avanzadas a laboratorios cotidianos
Para asegurar que esta tecnología sea utilizable más allá de los expertos en programación, los autores han integrado MS2DeepScore 2.0 en software de espectrometría de masas popular y de acceso libre. Con esta integración, los investigadores pueden detectar características, construir redes moleculares que ignoran las fronteras de modo de ionización y explorar el espacio químico resultante mediante paneles interactivos. El código, los modelos entrenados y los conjuntos de datos de ejemplo se comparten abiertamente, y el sistema puede volver a entrenarse o ajustarse para clases químicas especializadas.
Qué significa esto para futuros descubrimientos
Para los no especialistas, el mensaje clave es que MS2DeepScore 2.0 ayuda a convertir mediciones fragmentadas y dependientes del modo en una imagen única y más comprensible de las moléculas presentes en una muestra. Al vincular con fiabilidad señales que antes vivían en mundos analíticos separados, el método permite a los científicos aprovechar bibliotecas de referencia mucho más grandes, comparar muestras de forma más completa y centrar su atención en conjuntos significativos de compuestos relacionados. Se espera que esta interconexión de datos acelere la identificación de biomarcadores, nutrientes, productos naturales y contaminantes, profundizando en última instancia nuestra comprensión de cómo la química influye en la salud y el medio ambiente.
Cita: de Jonge, N.F., Chekmeneva, E., Schmid, R. et al. Cross ionization mode chemical similarity prediction between tandem mass spectra in metabolomics. Nat Commun 17, 2483 (2026). https://doi.org/10.1038/s41467-026-69083-y
Palabras clave: metabolómica, espectrometría de masas, aprendizaje automático, redes moleculares, similitud química