Clear Sky Science · es
Modelado de fibrosis pulmonar con iPSC humanas revela que la inhibición de p300/CBP suprime el estado transicional de las células alveolares
Por qué nos importan a todos los pulmones con cicatrices
La fibrosis pulmonar idiopática (FPI) es una enfermedad implacable en la que los pulmones se transforman lentamente en tejido cicatricial, haciendo que cada respiración sea más difícil. Los fármacos actuales solo pueden ralentizar este proceso y a menudo provocan efectos secundarios molestos. Este estudio emplea herramientas de vanguardia en células madre y genómica para recrear pulmones con cicatrices en el laboratorio, planteando una pregunta simple pero vital: ¿podemos encontrar un interruptor que desvía a las células pulmonares dañadas de un estado perjudicial y las vuelva a encaminar hacia la reparación?
Una ventana cultivada en laboratorio hacia un pulmón con cicatrices
Para investigar la FPI, los investigadores construyeron pulmones miniaturizados a partir de células madre pluripotentes inducidas humanas (iPSC). Estas iPSC se guiaron para convertirse en células alveolares —las que recubren los pequeños sacos de aire donde el oxígeno entra en la sangre— y se cultivaron junto con fibroblastos pulmonares, las células del tejido conectivo que forman la cicatriz. Embebidos en un gel blando, estos “organoides alveolares” se comportaron de manera muy similar al tejido pulmonar real. Al exponerse al fármaco quimioterápico bleomicina, un desencadenante conocido de lesión pulmonar, los geles se contrajeron a medida que los fibroblastos los tiraban, imitando la contracción tisular que se observa en la fibrosis.
Usando este sistema, el equipo cribó una biblioteca de 264 pequeñas moléculas y midió automáticamente cuánto evitaba cada fármaco la contracción del gel, con una herramienta de análisis de imágenes basada en aprendizaje profundo que garantizaba lecturas objetivas. Muchos compuestos no tuvieron efecto, pero una familia destacó de forma clara: los inhibidores de las proteínas p300 y CBP, que ayudan a controlar cómo se empaqueta el ADN y qué genes se activan. Las ocho moléculas dirigidas a p300/CBP de la biblioteca redujeron la contracción a bajas dosis, subrayando esta vía como un punto prometedor de intervención en la fibrosis. 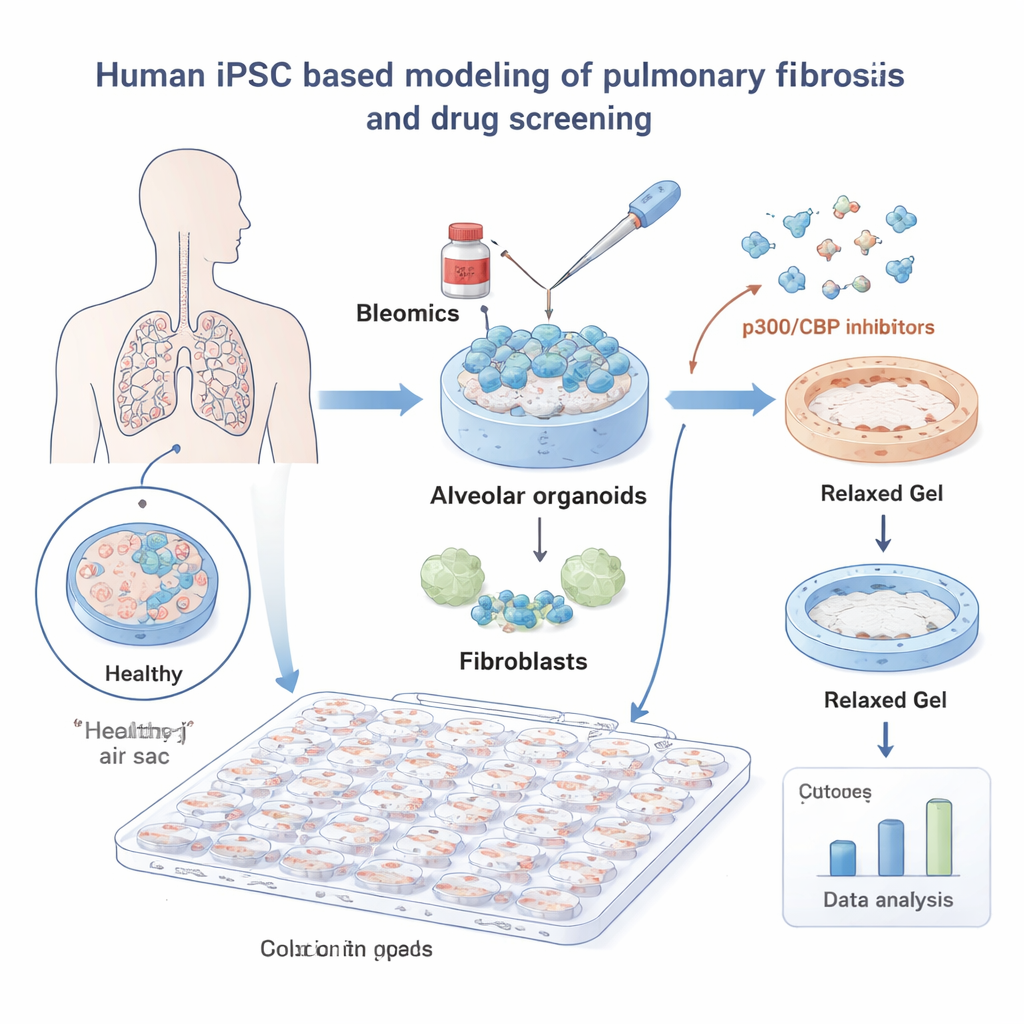
Los culpables: células pulmonares transicionales
Trabajos recientes han revelado un problemático tipo celular “intermedio” en pulmones enfermos, llamado estado transicional alveolar. Normalmente, las células de soporte conocidas como AT2 maduran hasta convertirse en las ultrafinas células AT1 que cubren los sacos de aire y posibilitan el intercambio gaseoso. En la FPI, sin embargo, las AT2 a menudo se quedan atascadas en este estado transicional, expresando genes de estrés y reparación mientras no completan la conversión a AT1 plenamente funcionales. Estas células transicionales se agrupan en regiones fibróticas y se comunican intensamente con los fibroblastos, pero no estaba claro si eran simplemente un subproducto del daño o motores activos de la cicatrización.
Al secuenciar ARN y perfilar la cromatina accesible en sus organoides, los autores mostraron que las células transicionales que surgían en su modelo coincidían estrechamente con las halladas en pulmones de pacientes con FPI. Estas células transicionales inducidas presentaban firmas génicas de estrés, inflamación y remodelado de matriz, y activaban fuertemente a los fibroblastos pulmonares cocultivados. De manera crucial, cuando se bloqueó p300/CBP, los marcadores del estado transicional disminuyeron, la identidad AT2 se preservó mejor y la activación de los fibroblastos se redujo. En otras palabras, los fármacos no envenenaban las células de forma generalizada; más bien, impedían selectivamente que las AT2 quedaran atrapadas en ese limbo perjudicial.
Desentrañando los interruptores moleculares
Para entender cómo p300/CBP modela esta decisión de destino, el equipo examinó marcas químicas en histonas —proteínas que ayudan a empaquetar el ADN. Una marca particular, la acetilación de H3K27, es depositada comúnmente por p300/CBP en potenciadores y promotores activos. En las células transicionales, regiones cercanas a genes de respuesta al estrés y pro‑fibrogénicos mostraban fuerte acetilación de H3K27 y estaban enriquecidas en sitios de unión de factores de transcripción como AP‑1 y HNF1B. Cuando las células se trataron con inhibidores de p300/CBP, estas marcas acetil se redujeron en esos sitios y la expresión de muchos genes pro‑fibrosis cayó. Bloquear AP‑1 directamente, o reducir AP‑1 y HNF1B mediante pequeños ARN interferentes, acortó asimismo el programa transicional y la contracción de los organoides, ligando a este trío —p300/CBP, AP‑1 y HNF1B— con el motor que impulsa el remodelado fibrótico.
Más allá de la placa, el estudio probó un inhibidor, CBP30, en ratones con lesión pulmonar inducida por bleomicina. Los animales tratados con CBP30 presentaron menos células epiteliales transicionales, menor activación de miofibroblastos formadores de cicatriz y expresión reducida de marcadores de fibrosis. Esta validación cruzada entre modelos humanos basados en células madre y un modelo animal refuerza la idea de que p300/CBP no es solo un artefacto de laboratorio sino un regulador genuino de la cicatrización pulmonar. 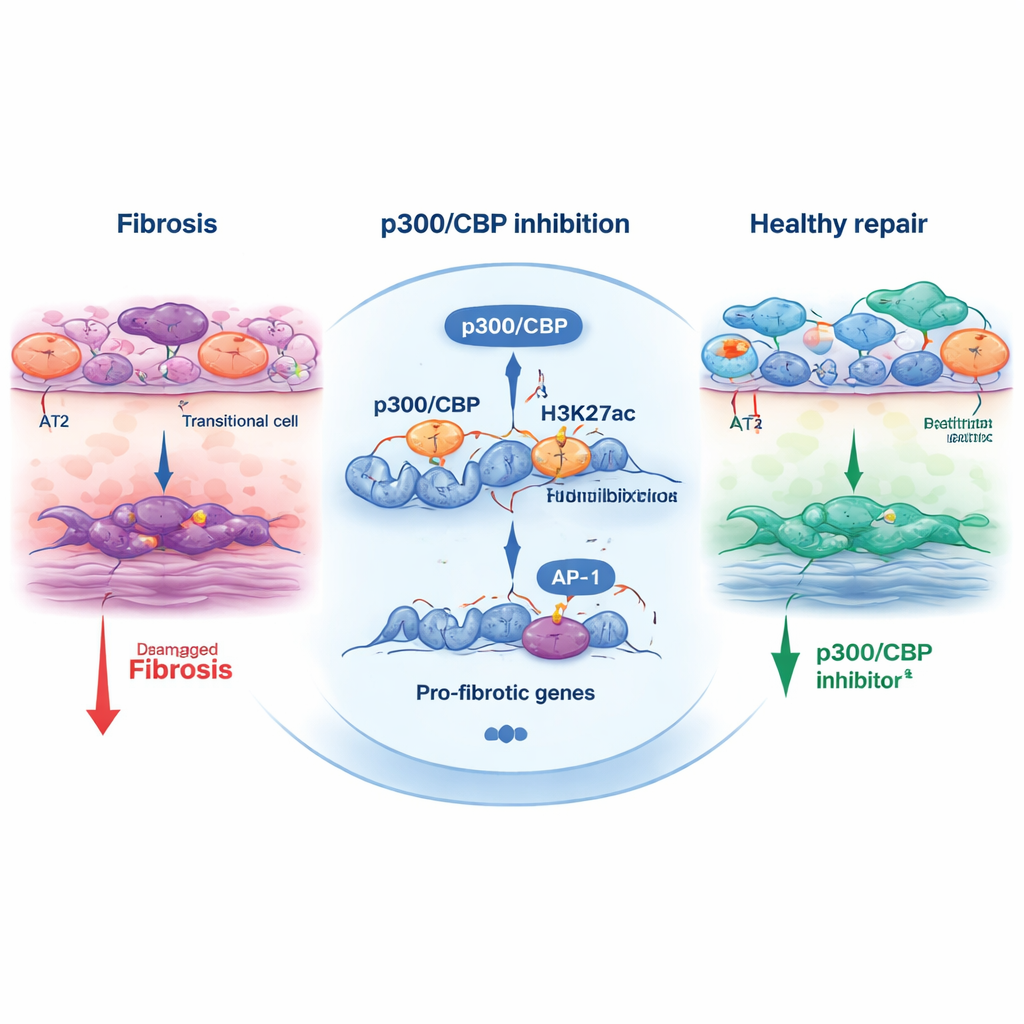
Qué significa esto para tratamientos futuros
Para el público general, la conclusión clave es que los autores han construido un modelo humano realista de pulmones fibróticos y lo han usado para señalar un nuevo objetivo terapéutico. Su trabajo sugiere que la cicatrización pulmonar está impulsada en parte por un estado transicional reversible inducido por estrés que desorienta al tejido circundante. Al reducir la actividad de p300/CBP, podría ser posible silenciar ese estado, mantener a las células alveolares en una trayectoria de desarrollo saludable y atenuar las señales que empujan a los fibroblastos a un estado de sobreactivación. Si bien los inhibidores de p300/CBP aún deben optimizarse en cuanto a seguridad y probarse clínicamente, este estudio apunta hacia terapias que abordan la descomunicación celular central en la FPI en lugar de limitarse a frenar sus consecuencias.
Cita: Tsutsui, Y., Masui, A., Konishi, S. et al. Human iPSC-based Modeling of Pulmonary Fibrosis Reveals p300/CBP Inhibition Suppresses Alveolar Transitional Cell State. Nat Commun 17, 1214 (2026). https://doi.org/10.1038/s41467-026-68909-z
Palabras clave: fibrosis pulmonar idiopática, organoides alveolares, inhibidores de p300/CBP, células epiteliales transicionales, células madre pulmonares