Clear Sky Science · es
El panorama funcional del splicing alternativo en la determinación de la línea hematopoyética
Cómo pequeños recortes en los genes moldean nuestra sangre
Cada segundo, su cuerpo produce millones de nuevas células sanguíneas. Detrás de este silencio milagro hay un sistema molecular de edición que puede cortar y pegar fragmentos de los mensajes genéticos de distintas maneras, generando versiones ligeramente diferentes de la misma proteína. Este estudio explora cómo ese proceso de edición, llamado splicing alternativo, ayuda a orientar las células madre inmaduras hacia convertirse en glóbulos rojos, glóbulos blancos u otros tipos de células sanguíneas —y qué ocurre cuando falta una sola pieza editada.
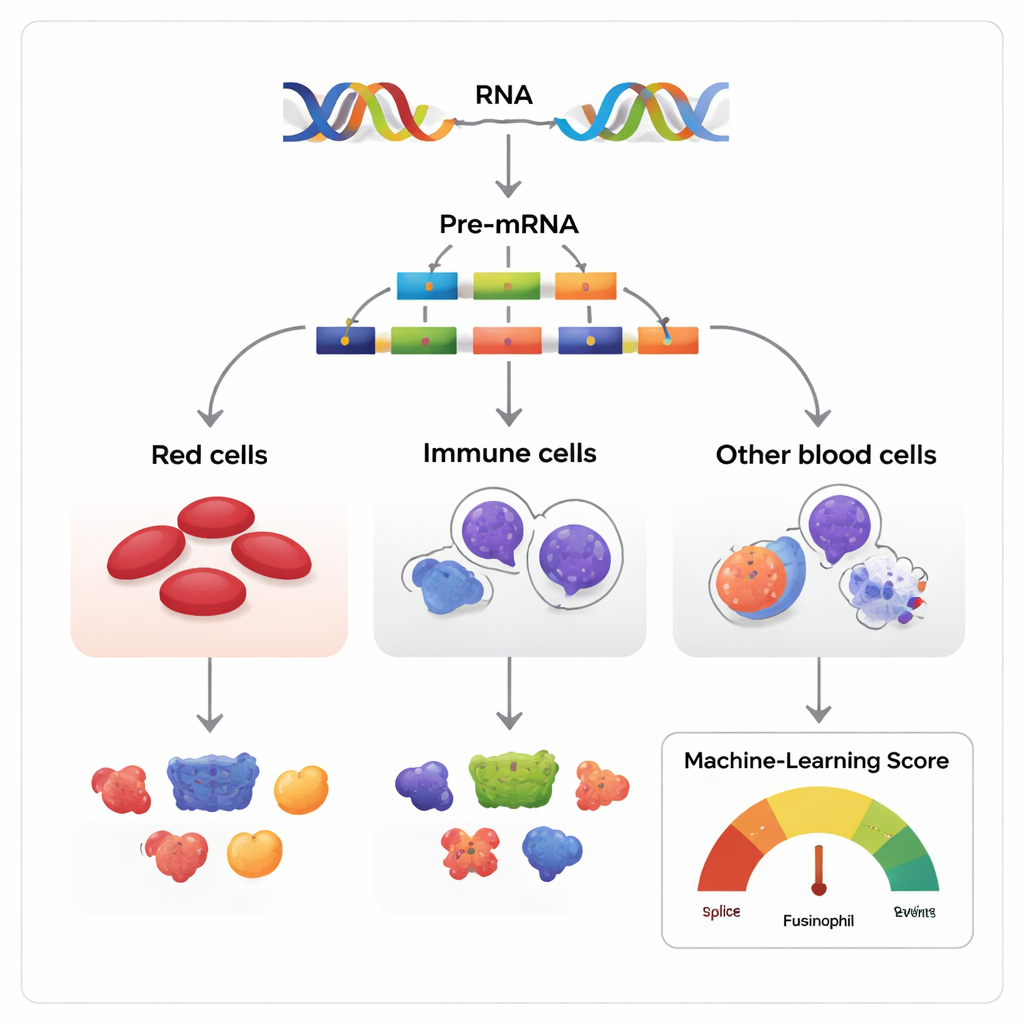
Muchas versiones a partir del mismo guion genético
Se suele describir a los genes como planos, pero en realidad se parecen más a guiones que pueden reordenarse. Cuando se lee un gen, la copia inicial de ARN contiene segmentos llamados exones que pueden conservarse o saltarse antes de que el mensaje final se traduzca en proteína. Los autores estudiaron este proceso, conocido como omisión de exones, en tejidos formadores de sangre de humanos, ratones y varios otros vertebrados. Reunieron más de 270 conjuntos de datos de ARN que siguen colectivamente a las células madre y progenitoras de la sangre mientras maduran hacia tres familias principales: las células eritroides productoras de glóbulos rojos, las células mieloides que combaten infecciones y las células linfoides que producen anticuerpos.
Clasificar qué cambios de splicing importan realmente
Dado que la mayoría de los genes con múltiples exones pueden empalmarse de muchas formas, el reto central es distinguir las variaciones inocuas de las que influyen de verdad en el destino celular. Los investigadores desarrollaron un modelo de aprendizaje automático, llamado Functional AS Score (FAScore), para abordar este problema. Para cada evento de omisión de exón, el modelo considera 19 piezas de información, como la intensidad del cambio de uso durante el desarrollo celular, el grado de conservación del ADN circundante entre especies, si altera dominios proteicos conocidos y si contiene sitios para modificaciones químicas de la proteína. El algoritmo, entrenado con una estrategia positive–unlabeled y un clasificador random forest, devuelve una puntuación entre 0 y 1 que indica la probabilidad de que un evento de empalme tenga un impacto funcional.
Detectar conmutadores conservados y específicos de linaje
Aplicando FAScore a decenas de miles de eventos de omisión de exones, el equipo los clasificó en grupos probables funcionales, no funcionales o inciertos. Los eventos predichos como funcionales se localizaron con más frecuencia en regiones proteicas importantes para interacciones, en secuencias evolutivamente conservadas y en fragmentos que alojan etiquetas químicas como fosforilación o SUMOilación. Muchos de estos eventos también estaban activos solo en linajes sanguíneos concretos o durante la formación fetal de la sangre, lo que sugiere que actúan como conmutadores finamente sintonizados en ventanas de desarrollo particulares. El estudio mostró además que algunos de los eventos de empalme más ancestrales —aquellos compartidos entre vertebrados durante cientos de millones de años— son especialmente propensos a ser funcionales, reflejando cómo los genes más antiguos tienden a conservar roles biológicos centrales.
Prueba experimental: ajustar los resultados en la formación celular sanguínea
Para poner a prueba las predicciones del modelo, los autores seleccionaron varios exones con puntuaciones altas en genes activos en distintos linajes sanguíneos y eliminaron esos exones en células madre y progenitoras sanguíneas de ratón mediante CRISPR. Los resultados concordaron con las expectativas del modelo: la eliminación de ciertos exones en los genes KLF6 y SSBP3 impidió la formación de colonias mieloides sin afectar la producción de glóbulos rojos, mientras que borrar exones en EPB41L1 y TBC1D23 alteró la formación de colonias eritroides. En particular, la omisión del exón 15 de TBC1D23 redujo la producción de precursores de glóbulos rojos en ratones y en peces cebra, provocando menos células rojas circulantes y niveles más bajos de hemoglobina, mientras que en gran medida preservó las células blancas.
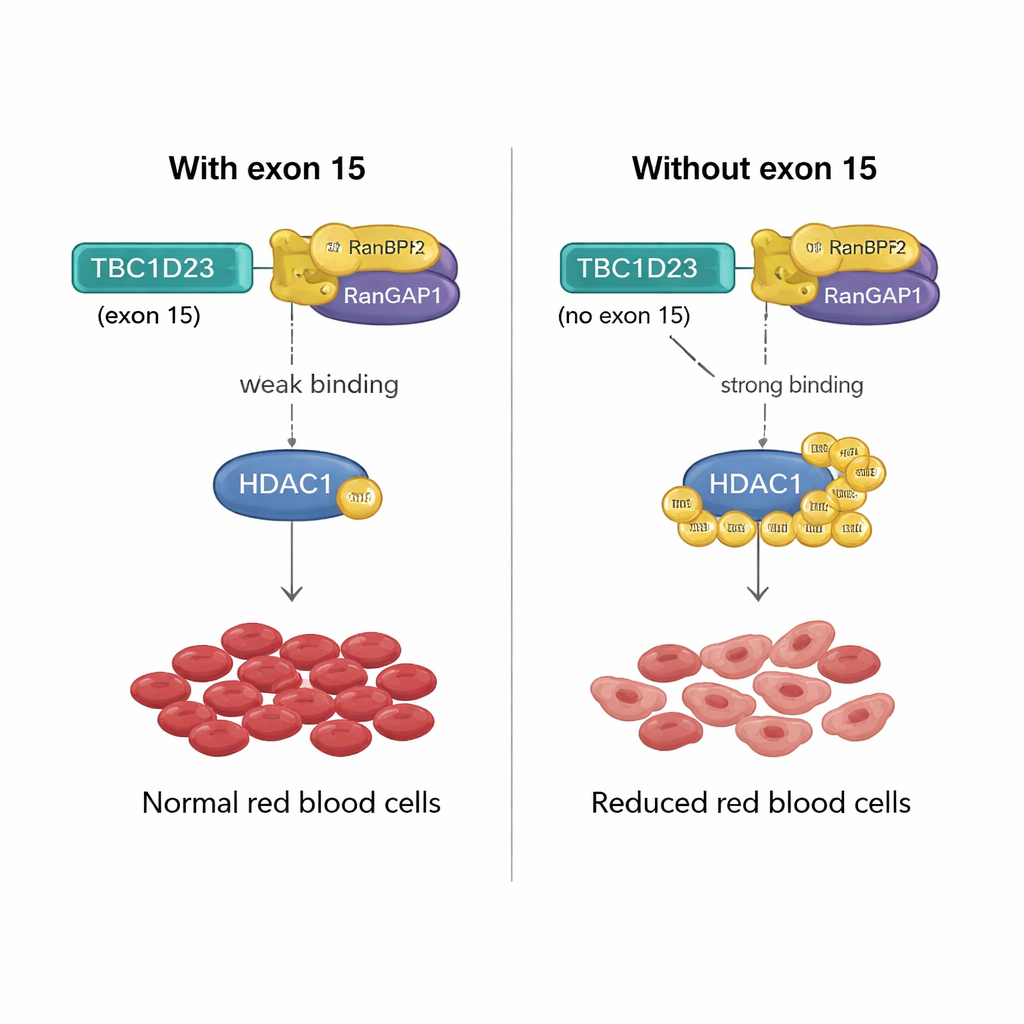
Una palanca molecular para la producción de glóbulos rojos
¿Cómo puede un tramo de 15 aminoácidos codificado por el exón 15 de TBC1D23 tener un efecto así? El equipo descubrió que la inclusión de este exón debilita la unión de TBC1D23 a un par de proteínas llamado RANBP2/RANGAP1, que colaboran para añadir etiquetas SUMO a otras proteínas. Sin el exón 15, TBC1D23 se une a este par con mayor fuerza, aumentando la SUMOilación de una enzima clave, HDAC1. Este marcado incrementado altera la actividad de muchos factores de transcripción —reguladores maestros de la expresión génica— desajustando los programas génicos necesarios para la maduración correcta de los glóbulos rojos. Cuando los investigadores diseñaron una versión de HDAC1 que no puede ser SUMOetiquetada en dos posiciones cruciales, rescataron la formación de glóbulos rojos en células sin el exón 15 de TBC1D23, confirmando que este marcado químico es el paso crítico.
Por qué importa esto para la salud y las terapias futuras
Para un no especialista, la lección de este trabajo es que no todos los cambios genéticos son iguales: a veces, la diferencia entre una sangre sana y una anemia reside en si un diminuto segmento de un gen se conserva o se omite en el mensaje final. Al combinar datos masivos de ARN con un sistema de puntuación sofisticado, el estudio ofrece una hoja de ruta para identificar qué variantes de empalme son más propensas a influir en cómo las células madre eligen su destino. Este enfoque no solo profundiza nuestra comprensión de cómo se forman las células sanguíneas en salud y enfermedad, sino que también propone una estrategia general para detectar eventos de splicing importantes en otros órganos, con el potencial de orientar futuras terapias génicas y tratamientos de precisión.
Cita: Hu, X., Wang, J., Chen, L. et al. The functional landscape of alternative splicing in hematopoietic lineage commitment. Nat Commun 17, 2047 (2026). https://doi.org/10.1038/s41467-026-68811-8
Palabras clave: splicing alternativo, hematopoyesis, aprendizaje automático, glóbulos rojos, regulación génica