Clear Sky Science · es
Predicción fiable de números de comisión de enzimas mediante un transformador jerárquico interpretable
Por qué importa predecir las funciones enzimáticas
Cada célula viva funciona gracias a innumerables pequeñas máquinas químicas llamadas enzimas. Cada enzima tiene un “trabajo” específico, y ese trabajo se codifica en un número de la Comisión de Enzimas (EC), un código de cuatro niveles parecido a una dirección postal. Asignar correctamente los números EC es crucial para entender el metabolismo, diseñar nuevos fármacos, diseñar microbios que produzcan combustibles o alternativas al plástico, y rastrear cómo los ecosistemas procesan los compuestos químicos. Pero los experimentos para determinar la función enzimática son lentos y caros. Este estudio presenta HIT-EC, un nuevo modelo de inteligencia artificial que puede predecir de forma fiable los números EC a partir de secuencias de proteínas y, además, explicar por qué realizó cada predicción.
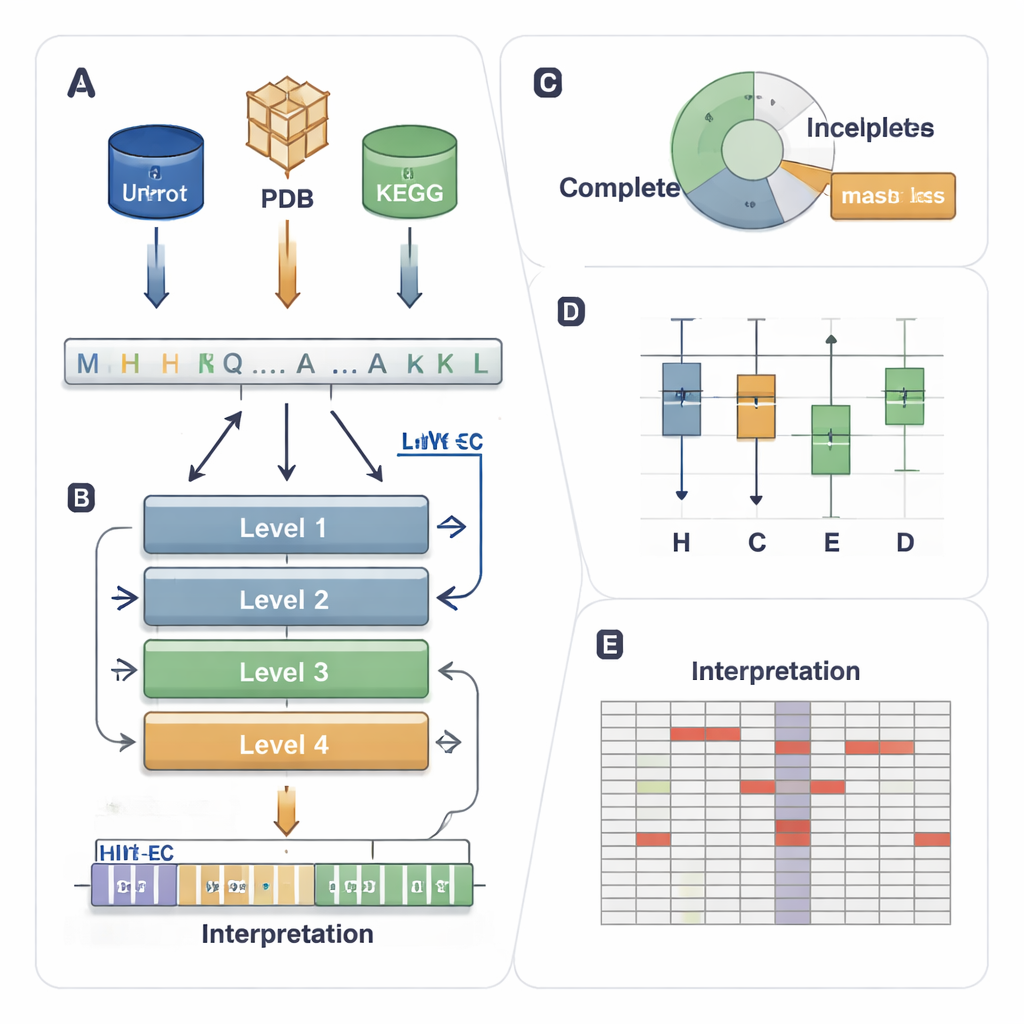
Un sistema tipo código postal para las funciones enzimáticas
El sistema EC asigna a cada enzima un código de cuatro niveles como 1.1.1.37. El primer dígito indica una clase amplia (por ejemplo, enzimas que mueven electrones o transfieren grupos), y los dígitos posteriores describen detalles más precisos de la reacción. Esta jerarquía es poderosa pero plantea un problema de predicción exigente: un modelo debe acertar los cuatro niveles para miles de códigos posibles, incluso cuando algunas enzimas son raras o están anotadas solo parcialmente en las bases de datos (por ejemplo, 3.5.-.-, donde faltan niveles detallados). Los métodos informáticos existentes usan estructura 3D, similitud de secuencia o aprendizaje profundo, pero suelen tener dificultades con enzimas poco frecuentes, ignoran datos parcialmente etiquetados y, por lo general, actúan como “cajas negras” que ofrecen poca información sobre por qué tomaron una decisión.
Una IA de cuatro pisos que sigue la escalera EC
HIT-EC (Hierarchical Interpretable Transformer for EC prediction) está diseñado para reflejar la jerarquía de cuatro pasos del sistema EC. Toma una secuencia de proteína sin procesar y la pasa por cuatro capas de transformador, cada una centrada en un nivel EC. Flujos locales conectan cada nivel con el anterior, asegurando que una decisión fina (el cuarto dígito) sea consistente con las más generales (primer y segundo dígito). En paralelo, un flujo global mantiene el contexto completo de la secuencia visible en cada paso. El modelo también puede entrenarse con secuencias con etiquetas incompletas, usando una “pérdida enmascarada” que simplemente ignora los niveles EC faltantes en lugar de descartar la secuencia. Esto permite que HIT-EC aprenda de la gran fracción de proteínas en bases de datos curadas que están solo parcialmente anotadas.
Superando a rivales en precisión y velocidad
Los autores reunieron un conjunto de datos grande y cuidadosamente filtrado de aproximadamente 200 000 enzimas con 1 938 números EC diferentes procedentes de Swiss-Prot y del Protein Data Bank. En pruebas de retención repetidas, HIT-EC superó a tres métodos líderes (CLEAN, ECPICK y DeepECtransformer) tanto en F1 global como por clase, que miden el equilibrio entre aciertos correctos y falsos positivos. Fue especialmente fuerte en códigos EC poco representados con 25 o menos ejemplos conocidos, donde los métodos anteriores suelen fallar. HIT-EC también generalizó bien a enzimas nuevas añadidas a Swiss-Prot tras el entrenamiento y a genomas completos de bacterias diversas, incluidas cepas bien estudiadas de Escherichia coli, Bacillus subtilis y Mycobacterium tuberculosis. A pesar de su sofisticación, el modelo resultó muy eficiente: en una GPU estándar procesó una proteína en alrededor de 38 milisegundos, decenas de veces más rápido que algunos competidores que dependen de búsquedas de similitud más lentas o de conjuntos de muchos modelos.
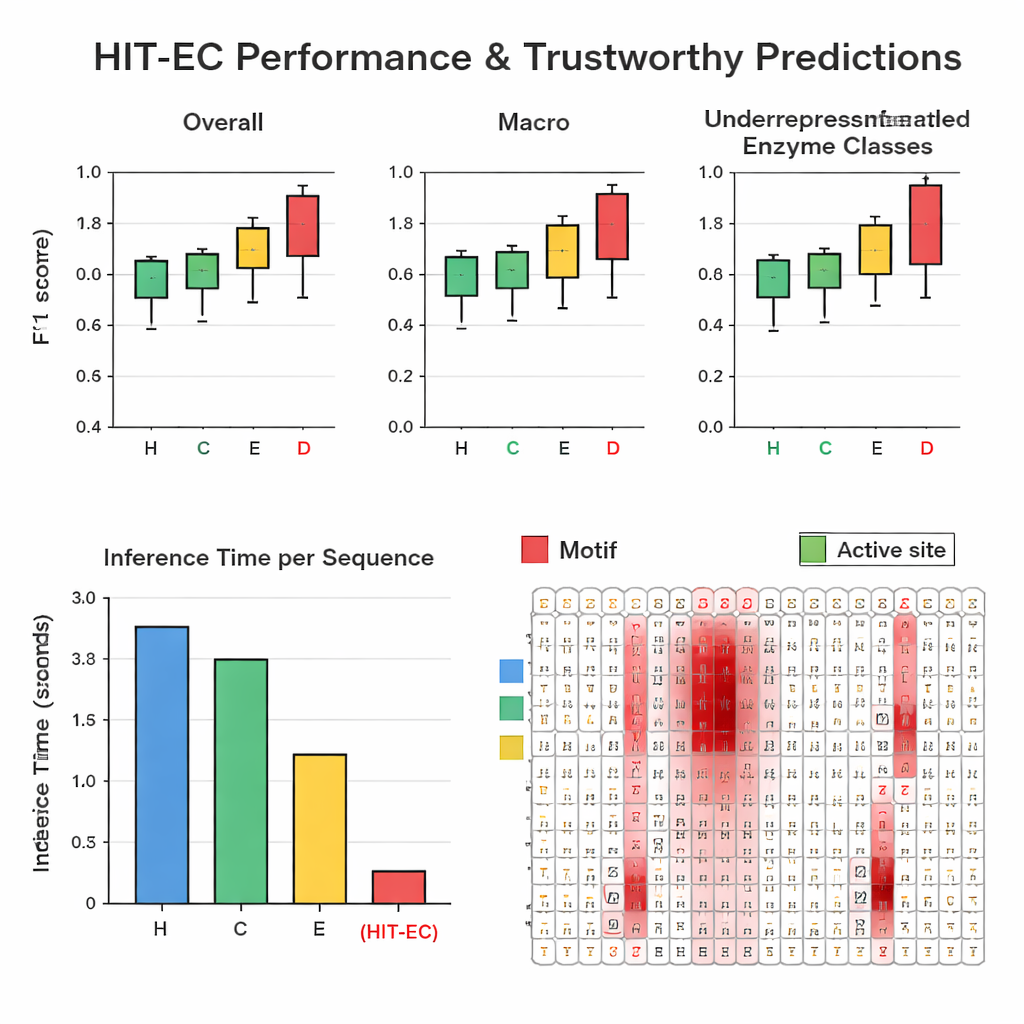
Ver en qué “mira” el modelo
Para que sus predicciones sean confiables, HIT-EC está diseñado para mostrar qué aminoácidos de la secuencia influyeron en cada decisión por nivel EC. Los autores construyeron una vía de interpretación que combina pesos de atención con información de gradiente para puntuar la importancia de cada posición. Validaron estas puntuaciones en familias de enzimas bien caracterizadas. Por ejemplo, en una familia de citocromo P450 (CYP106A2), HIT-EC destacó motivos funcionales conocidos como regiones de unión al oxígeno y al grupo hemo, e identificó un sutil motivo EXXR que un modelo de referencia pasó por alto. Para representantes clásicos de cada clase EC superior —como alcohol deshidrogenasa, hexocinasa y anhidrasa carbónica—, las puntuaciones de relevancia del modelo activaron motivos distintivos de libro de texto y sitios de unión al sustrato. Estas interpretaciones proporcionan “evidencia” bioquímica de que el modelo basa sus predicciones en características significativas y no en correlaciones accidentales.
Orientando el trabajo sobre enzimas raras y emergentes
El equipo probó además HIT-EC en dos enzimas poco estudiadas importantes para la limpieza de contaminantes: un citocromo P450 implicado en la degradación de contaminantes aromáticos y una hidrolasa degradadora de PET de Streptomyces que ayuda a digerir moléculas relacionadas con el plástico. Ambas enzimas habían sido caracterizadas experimentalmente pero carecían de asignaciones EC oficiales. HIT-EC predijo correctamente los números EC esperados y destacó patrones de motivos y residuos catalíticos que coinciden con lo conocido a partir de estudios estructurales y bioquímicos. En conjunto, el trabajo muestra que HIT-EC no solo puede asignar números EC con mayor precisión y rapidez que las herramientas actuales, especialmente para funciones raras, sino también esclarecer por qué se cree que una enzima realiza una tarea química determinada. Esta combinación de rendimiento e interpretabilidad lo convierte en un motor prometedor para una anotación enzimática a gran escala, fiable, en genómica, biotecnología e investigación ambiental.
Cita: Dumontet, L., Han, SR., Lee, J.H. et al. Trustworthy prediction of enzyme commission numbers using a hierarchical interpretable transformer. Nat Commun 17, 1146 (2026). https://doi.org/10.1038/s41467-026-68727-3
Palabras clave: predicción de función enzimática, aprendizaje profundo en biología, modelos transformadores, anotación de proteínas, enzimas para biorremediación