Clear Sky Science · es
Una activación alternativa de EGFR por la mutación R252C derivada de un paciente promueve la progresión del cáncer
Cuando las antenas celulares se rebelan
¿Por qué algunos cánceres siguen creciendo incluso tras rondas de quimioterapia e inmunoterapias de última generación? Este estudio sigue a un paciente con tumores en pulmón y cerebro y rastrea su enfermedad hasta un pequeño cambio en una “antena” clave de la superficie celular llamada EGFR. Al descubrir cómo esta única mutación reconfigura las señales de crecimiento, los investigadores no solo explican el carácter agresivo del cáncer del paciente, sino que también muestran cómo un fármaco existente, el afatinib, puede controlarlo.
Una mutación rara con grandes consecuencias
EGFR es un receptor que atraviesa la membrana celular y ayuda a las células a responder a señales de crecimiento. Muchos cánceres de pulmón y cerebro presentan mutaciones en EGFR, pero la mayoría de las alteraciones conocidas se localizan en el interior de la célula, en la porción que actúa como un interruptor enzimático. Aquí, el equipo descubrió un cambio inusual en el exterior de EGFR, en la parte que normalmente captura factores de crecimiento. En un paciente con cáncer de pulmón y glioma, hallaron que un aminoácido en la posición 252 se había sustituido: de arginina a cisteína —denominado EGFR R252C. La minería de bases de datos de cáncer mostró esta mutación en una pequeña fracción de pacientes con glioma y casi nunca en tumores de pulmón, lo que sugiere que es rara pero real. Usando herramientas de edición genética, los autores recrearon exactamente esta mutación en varias líneas celulares humanas de cáncer cerebral y pulmonar para probar su efecto.
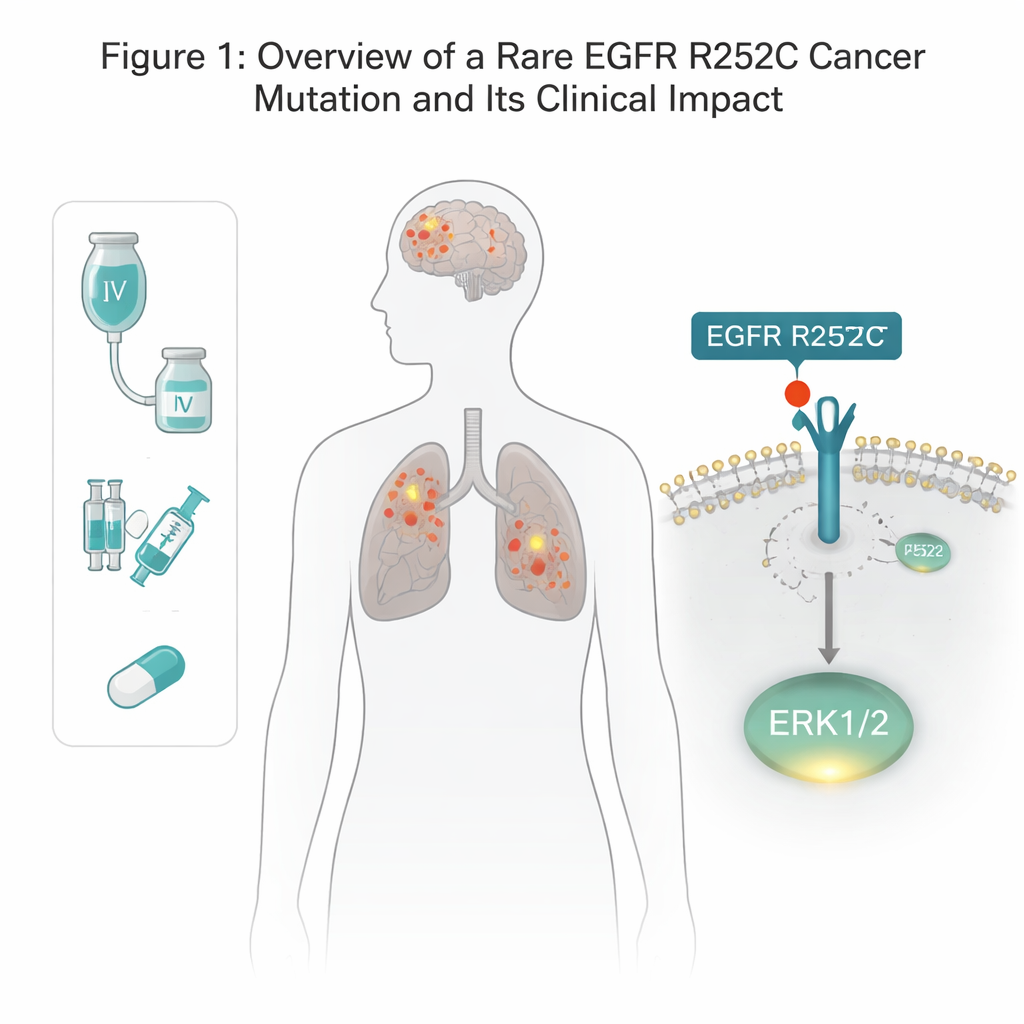
Un nuevo atajo hacia las señales de crecimiento
Normalmente, EGFR debe emparejarse con otra copia y luego marcar su propia cola interior con grupos fosfato antes de poder activar las vías de crecimiento aguas abajo. Sorprendentemente, la versión R252C no mostró ese autocontrol habitual. En cambio, las células que portaban EGFR R252C activaron de manera mucho más intensa a un controlador de crecimiento específico, ERK1/2, mientras que otras vías clásicas de EGFR —como AKT y STAT3— quedaron en gran medida sin cambios. Bloquear ERK1/2 con un inhibidor específico eliminó la ventaja de crecimiento adicional de las células R252C, demostrando que ERK1/2 es el motor principal detrás del poder tumorigénico de esta mutación.
Bloquear el receptor en una forma siempre activa
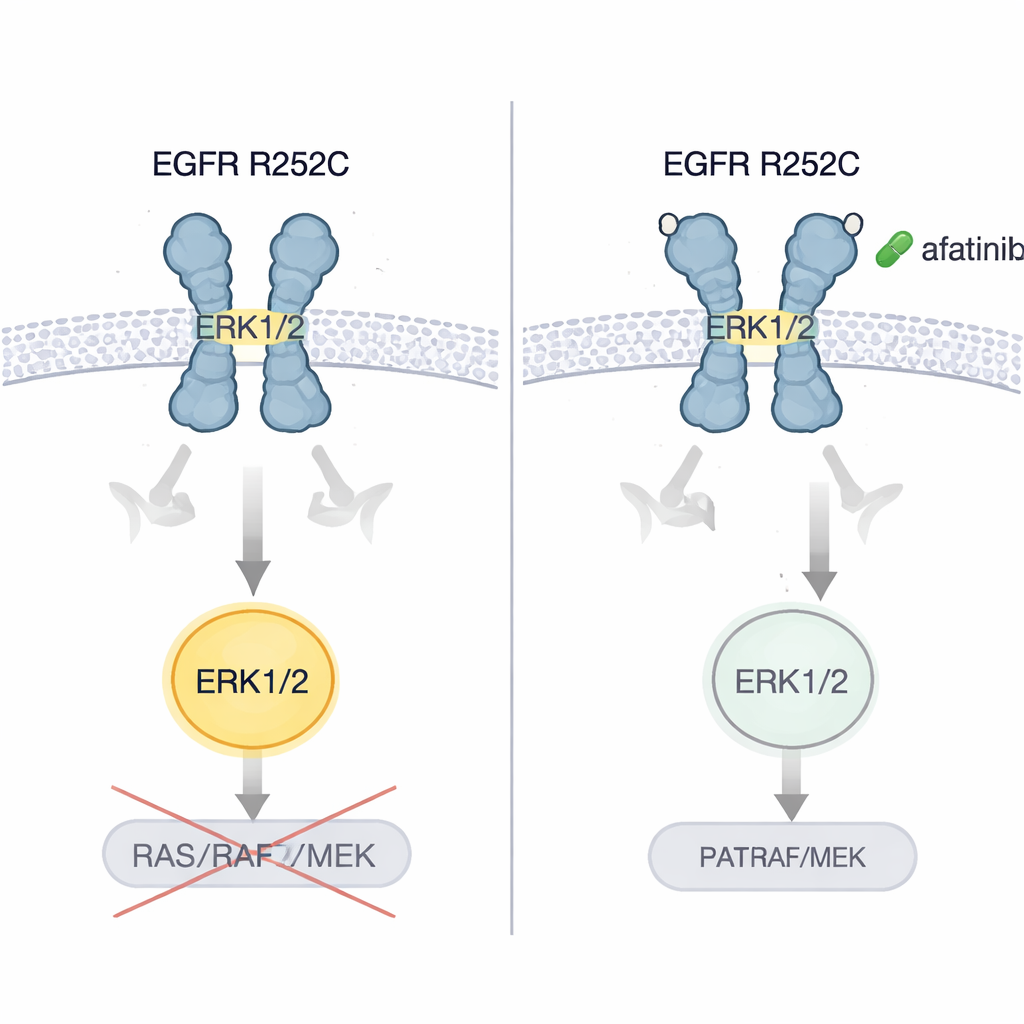
Para entender cómo un cambio externo podía provocar una sobreactivación tan selectiva, los investigadores combinaron ensayos bioquímicos con simulaciones por ordenador. El intercambio R252C introduce una nueva cisteína en la porción exterior de EGFR. Dos de esos mutantes pueden formar un enlace disulfuro —una especie de grapa molecular— entre sus residuos C252, uniéndolos en un par estable. El modelado estructural mostró que este enlace fuerza la parte exterior del receptor en una alineación escalonada en forma de “V” que imita de cerca la forma activa ligada al ligando, incluso sin presencia de factor de crecimiento. Esta alineación se propaga a través de los segmentos que atraviesan la membrana y los inmediatos al interior, torsionando los dominios enzimáticos internos en una disposición inusual: los sitios activos miran hacia el interior de la célula pero quedan demasiado separados para marcarse entre sí con eficacia. En cambio, esta conformación crea una superficie de acoplamiento fuerte para ERK1/2, permitiendo que EGFR R252C fosforile directamente a ERK1/2 y eluda el relevo clásico RAS–RAF–MEK.
De modelos en ratón a un solo paciente
Los autores demostraron que las células de cáncer cerebral y pulmonar que portaban EGFR R252C crecían más rápido en cultivo y formaban tumores más grandes y agresivos al implantarlas en ratones, en comparación con células que llevaban EGFR normal. Luego probaron varias generaciones de inhibidores orales de EGFR. Solo el afatinib, un inhibidor de segunda generación, apagó de forma consistente la activación de ERK1/2 y redujo drásticamente el crecimiento de las células tumorales. En modelos murinos de tumores cerebrales y pulmonares impulsados por R252C, el afatinib ralentizó la expansión tumoral y prolongó la supervivencia. De manera crítica, cuando el paciente original —cuya enfermedad había empeorado a pesar de quimioterapia, un fármaco dirigido a los vasos sanguíneos y la inmunoterapia— fue tratado con afatinib, las exploraciones de pulmón y cerebro mostraron una reducción notable de la carga tumoral y el paciente disfrutó de varios años sin progresión.
Qué significa esto para los pacientes
Este trabajo revela una forma hasta ahora no reconocida en que una mutación oncogénica de EGFR puede operar: grapando dos receptores fuera de la célula, torciéndolos en una pose activa que enciende directamente a ERK1/2 en lugar de seguir la cadena de señalización clásica. Para el público general, la conclusión clave es que no todas las mutaciones en un mismo gen se comportan igual, y algunos cambios raros pueden responder mejor a fármacos existentes concretos. EGFR R252C parece generar cánceres particularmente vulnerables al afatinib. Aunque esta conclusión se basa actualmente en un caso clínico detallado y en abundante trabajo de laboratorio, apunta hacia pruebas más personalizadas de las mutaciones del dominio externo de EGFR y sugiere que terapias dirigidas elegidas con criterio podrían ofrecer nueva esperanza a pacientes selectos con tumores cerebrales y pulmonares difíciles de tratar.
Cita: Zhang, Y., Fei, Q., Li, Y. et al. An alternative EGFR activation by patient-derived R252C mutation promotes cancer progression. Nat Commun 17, 1902 (2026). https://doi.org/10.1038/s41467-026-68699-4
Palabras clave: mutación EGFR, glioma, cáncer de pulmón, señalización ERK, afatinib