Clear Sky Science · es
Estimación de la estabilidad del plegamiento de proteínas con consideración explícita de los estados desplegados
Por qué importa la estabilidad de las proteínas
Cada proteína de tu cuerpo es una pequeña máquina molecular que debe plegarse en una forma tridimensional precisa para funcionar correctamente. Si ese plegamiento es demasiado frágil, la proteína puede fallar en su función, agregarse con otras proteínas o no llegar a producirse en absoluto, problemas que se vinculan con enfermedades y con fracasos en la fabricación de fármacos y enzimas basados en proteínas. Medir cuán estable es una proteína en el laboratorio es lento y complejo, por lo que los científicos buscan métodos computacionales que puedan indicar de forma fiable, a partir de la secuencia sola, con qué facilidad una proteína se desplegará.
Una nueva mirada a las proteínas plegadas y desplegadas
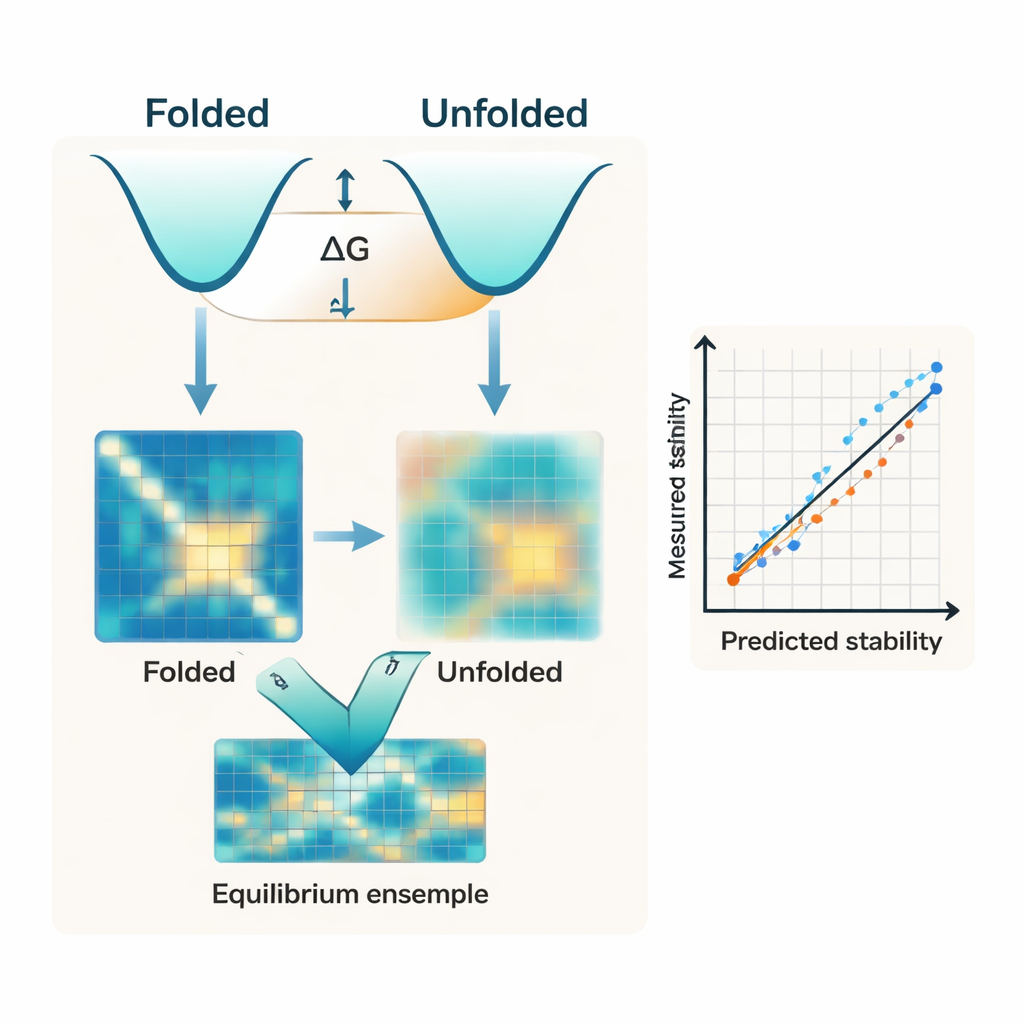
La mayoría de los algoritmos modernos se centran casi exclusivamente en la forma plegada de una proteína. A menudo parten de una estructura predicha por IA, como las de AlphaFold, y tratan esa única estructura como el determinante principal de la estabilidad. Pero la estabilidad es en realidad la diferencia de energía entre dos conjuntos amplios: el estado compacto plegado y las muchas formas flexibles que componen el estado desplegado. Los autores sostienen que ignorar el lado desplegado de este equilibrio es una razón clave por la que las herramientas existentes tienen dificultades para coincidir con las medidas experimentales de la energía libre de plegamiento, conocida como ΔG.
Un nuevo modelo que aprende ambos estados
Los investigadores presentan IFUM, un sistema de aprendizaje profundo diseñado para estimar ΔG al tiempo que aprende cómo es el equilibrio entre estados plegados y desplegados para cada proteína. En lugar de tratar el estado desplegado como un fondo vago, IFUM utiliza ideas de la física de polímeros para representarlo como una “bobina aleatoria” y codifica tanto los estados plegado como desplegado como mapas de distancias entre pares de aminoácidos. El modelo incorpora información de potentes redes preentrenadas de secuencia y estructura, y predice de forma conjunta la estabilidad total y un mapa de probabilidades que describe qué fracción de la población de proteínas está plegada frente a desplegada en cada par de residuos. El entrenamiento con un conjunto de datos muy grande de proteínas pequeñas caracterizadas experimentalmente y de proteínas desordenadas conocidas ayuda a IFUM a reconocer tanto secuencias bien estructuradas como flexibles.
Números mejores y mayor cobertura de mutaciones
Al evaluarse en un conjunto de datos cuidadosamente controlado de proteínas pequeñas, IFUM predice los valores experimentales de ΔG con menor error y mayor correlación que métodos basados en IA previos que dependen solo de la estructura plegada o de modelos de lenguaje entrenados en secuencias. De forma crucial, el modelo también maneja una amplia variedad de cambios en la secuencia. Captura con precisión los efectos de mutaciones puntuales simples y dobles, así como inserciones y deleciones que cambian la longitud de la proteína, situaciones en las que muchas herramientas existentes fallan por completo o no están diseñadas para operar. Una comparación interna muestra que eliminar el objetivo relativo al estado desplegado empeora significativamente el rendimiento, lo que subraya que modelar explícitamente el conjunto desplegado no es solo una nicetía conceptual, sino central para la precisión de las predicciones.
Del banco de diseño a pruebas en el mundo real
Para comprobar si IFUM puede guiar la ingeniería real de proteínas, los autores lo aplican a tres problemas de diseño desafiantes: estabilizar proteínas interferón‑lambda, remodelar la proteína de señalización inmunitaria IL‑10 y mejorar una enzima modificadora de azúcares llamada UGT76G1. En los tres casos, las estabilidad previstas por IFUM se correlacionan bien con las temperaturas de fusión medidas, que indican cuánta temperatura puede soportar una proteína antes de desplegarse. El modelo también ayuda a cribar cientos de proteínas completamente nuevas diseñadas por ordenador para seleccionar aquellas con mayor probabilidad de plegarse y permanecer solubles en células, superando a las puntuaciones de confianza ampliamente usadas de las redes de predicción de estructuras. Estos resultados sugieren que IFUM puede usarse como un “filtro de estabilidad” práctico junto a las comprobaciones basadas en estructura en los flujos de trabajo modernos de diseño de proteínas.
Límites y direcciones futuras
Como cualquier modelo, IFUM tiene límites. Está entrenado principalmente con proteínas cortas, de una sola cadena y solubles, y sus cifras absolutas de estabilidad pierden fiabilidad para proteínas mucho más grandes o para aquellas con lazos flexibles extensos o regiones que atraviesan membranas. Su descripción del estado desplegado sigue siendo un modelo estadístico simplificado más que una imagen completamente realista de todas las formas posibles. No obstante, el enfoque demuestra que enseñar a una IA a considerar ambos conjuntos, plegado y desplegado, proporciona estimaciones de estabilidad más fiables. Para los no expertos, la conclusión clave es que IFUM nos acerca a poder preguntar a un ordenador, con confianza cuantitativa, «¿Este diseño de proteína realmente se mantendrá unido?», lo que podría acelerar el desarrollo de fármacos biológicos más seguros y enzimas industriales más robustas.
Cita: Lee, H., Cho, Y., Yun, J. et al. Protein folding stability estimation with explicit consideration of unfolded states. Nat Commun 17, 1883 (2026). https://doi.org/10.1038/s41467-026-68637-4
Palabras clave: estabilidad de proteínas, plegamiento de proteínas, aprendizaje profundo, diseño de proteínas, mutaciones