Clear Sky Science · es
Un modelo de lenguaje genómico mitiga artefactos de quimeras en la secuenciación directa de ARN por nanoporo
Por qué importa depurar las lecturas de ARN
Nuestras células leen constantemente instrucciones genéticas escritas en ARN, y las nuevas tecnologías de secuenciación permiten ahora a los científicos observar este proceso con un detalle sin precedentes. Una de las herramientas más potentes, la secuenciación directa de ARN por nanoporo, puede leer moléculas de ARN completas de una sola pasada, pero también introduce fallos que pueden hacer que parezca que los genes están rotos y reensamblados de formas que no ocurren en la realidad. Este estudio presenta DeepChopper, una herramienta de software que actúa como un modelo de lenguaje para genomas, limpiando estos errores para que los investigadores puedan confiar en lo que ven en los datos de ARN.
Cuando el secuenciador inventa mezclas génicas falsas
Las máquinas modernas de nanoporo hacen pasar hebras individuales de ARN a través de poros diminutos y leen su secuencia directamente. Esto tiene grandes ventajas sobre métodos anteriores, como preservar modificaciones químicas y capturar transcritos de longitud completa en una sola lectura. Pero el proceso también depende de pequeños fragmentos auxiliares llamados adaptadores que se pegan a las moléculas de ARN durante la preparación de la librería. A veces, dos o más moléculas de ARN se unen por accidente mediante estos adaptadores, creando lo que parecen quimeras: moléculas híbridas que parecen fusionar genes distintos. Las herramientas de análisis estándar pueden malinterpretar estos restos técnicos como eventos biológicos reales, como fusiones génicas relacionadas con el cáncer o patrones de empalme inusuales, lo que conduce a resultados engañosos.
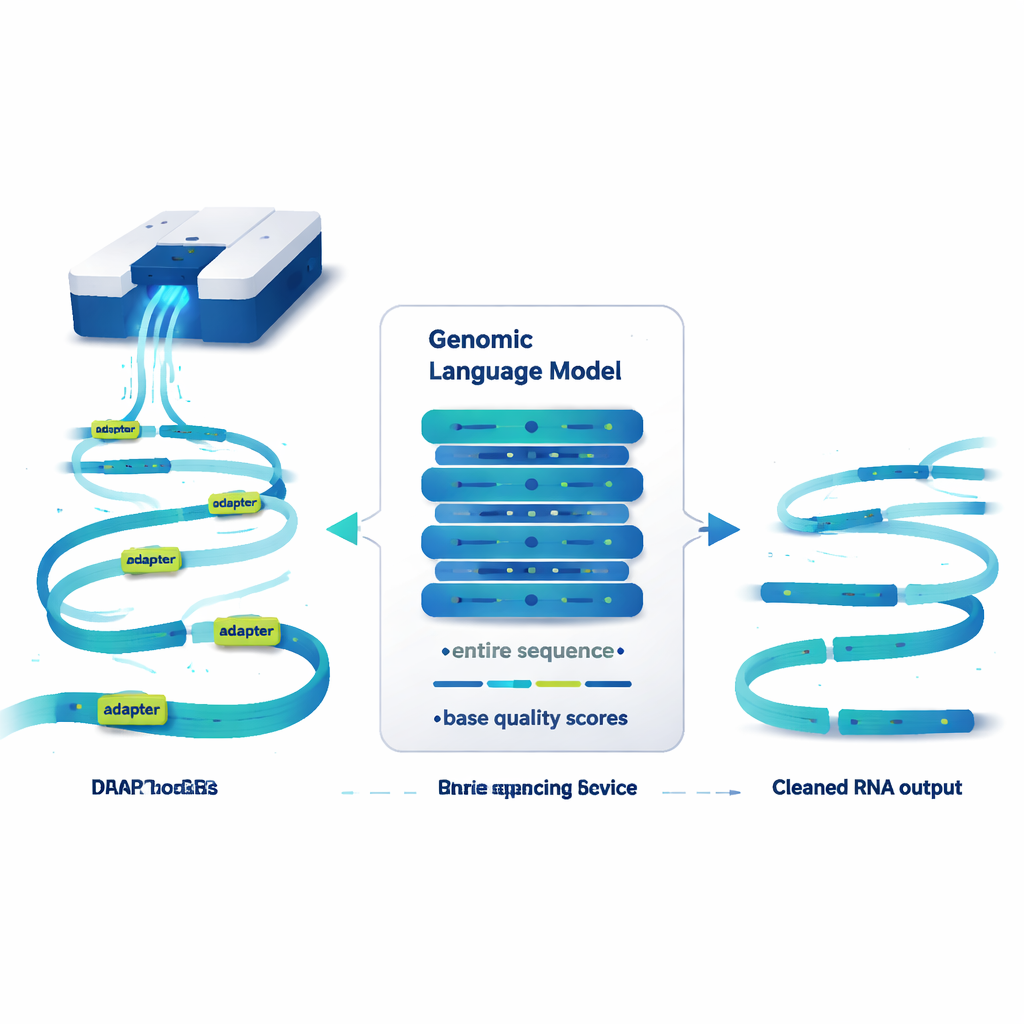
Un modelo de lenguaje que lee genomas, no oraciones
DeepChopper trata las secuencias genéticas un poco como texto y aplica ideas de los grandes modelos de lenguaje a ellas. En lugar de trabajar con palabras, lee secuencias de ARN letra por letra, junto con una puntuación de calidad para cada letra que indica cuán fiable es la lectura. Construido sobre una arquitectura compacta llamada HyenaDNA, puede escanear hasta 32.000 bases a la vez, lo suficiente para cubrir prácticamente cualquier molécula de ARN humana. Para cada posición, DeepChopper estima si esa base forma parte de una secuencia de ARN genuina o de un adaptador. Un paso de refinamiento luego suaviza esas predicciones para que los adaptadores se marquen como bloques continuos en lugar de puntos dispersos.
Cortar las uniones malas sin desechar datos
Una vez que DeepChopper ha encontrado adaptadores dentro de una lectura, hace algo crucial: en lugar de descartar toda la lectura, «corta» en esos sitios de adaptador y conserva las piezas reales. De este modo, una fusión artificial de dos ARNs puede dividirse de nuevo en sus partes originales. En pruebas con millones de lecturas por nanoporo procedentes de varias líneas celulares humanas de cáncer y de células madre, DeepChopper superó con creces a las herramientas existentes de recorte de adaptadores, que nunca fueron diseñadas para este contexto de ARN directo. Reconoció adaptadores con más del 99 % de precisión y exhaustividad en bancos de pruebas sintéticos, y escaló de forma eficiente a conjuntos de datos con más de 20 millones de lecturas utilizando procesadores gráficos.
Separar fusiones génicas reales de espejismos de secuenciación
Los autores preguntaron entonces si DeepChopper podía distinguir eventos biológicos genuinos de artefactos en datos reales de cáncer. Comparando lecturas directas de ARN con conjuntos de datos coincidentes producidos por métodos independientes (como la secuenciación directa de ADN complementario en plataformas de Oxford Nanopore y PacBio), pudieron etiquetar qué quimeras aparentes estaban respaldadas por otras tecnologías y cuáles no. DeepChopper redujo las alineaciones quiméricas no respaldadas hasta en un 62–91 %, mientras enriquecía considerablemente la fracción confirmada por otros métodos. También redujo el número de llamadas sospechosas de fusiones génicas en casi un 90 %, especialmente las que involucraban genes ribosomales que resultaron ser artefactos frecuentes. Al mismo tiempo, los eventos de fusión verdaderos respaldados por secuenciación de lecturas cortas se preservaron.
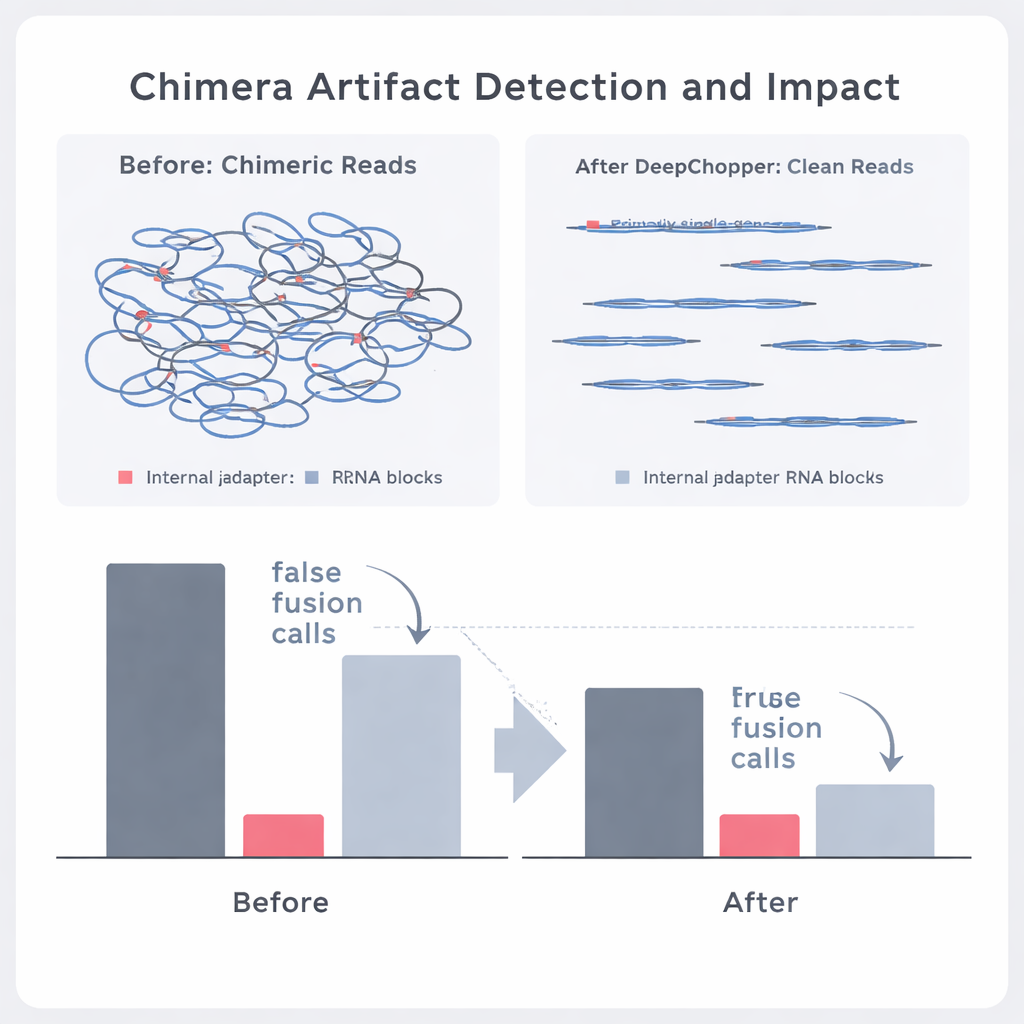
Mejor química ayuda, pero los artefactos persisten
Oxford Nanopore lanzó recientemente un kit de secuenciación actualizado (RNA004) diseñado en parte para reducir artefactos técnicos. DeepChopper se aplicó inicialmente «tal cual» a datos de esta nueva química y aun así encontró que una fracción pequeña pero importante de lecturas contenía adaptadores internos y uniones quiméricas. Incluso sin entrenamiento adicional, el modelo redujo las quimeras artefactuales en alrededor de una quinta parte; tras un ajuste fino con los nuevos datos, rindió algo mejor, mientras mantenía intactas las señales genuinas. En todas las químicas y tipos celulares, corregir estos artefactos permitió a las herramientas posteriores detectar muchas más transcripciones de longitud completa y alternativas, ofreciendo una visión más clara del paisaje de ARN de la célula.
Qué significa esto para futuros estudios de ARN
Para no especialistas, el mensaje clave es que no toda conexión sorprendente de ARN reportada por un secuenciador es biología real: algunas son errores de cableado introducidos por la propia tecnología. DeepChopper actúa como un corrector altamente entrenado para datos de ARN por nanoporo, detectando las secuencias de adaptador que unen moléculas no relacionadas y cortándolas con precisión de una sola base. El resultado son mapas más limpios y fiables de qué moléculas de ARN existen en una célula y cómo están compuestas. A medida que los laboratorios dependan cada vez más de la secuenciación de ARN de lecturas largas para estudiar cáncer, trastornos neurológicos y otras enfermedades complejas, herramientas como DeepChopper serán esenciales para convertir lecturas ruidosas en conocimientos biológicos fiables.
Cita: Li, Y., Wang, TY., Guo, Q. et al. Genomic language model mitigates chimera artifacts in nanopore direct RNA sequencing. Nat Commun 17, 1864 (2026). https://doi.org/10.1038/s41467-026-68571-5
Palabras clave: secuenciación de ARN por nanoporo, lecturas quiméricas, artefactos de fusión génica, modelo de lenguaje genómico, DeepChopper