Los cánceres de pulmón resistentes a inhibidores de EGFR muestran sensibilidad colateral a un puente molecular covalente y oligomerizante de KEAP1 independiente de cisteína
Por qué importa el cáncer de pulmón resistente a fármacos
Los fármacos dirigidos han transformado el tratamiento de ciertos cánceres de pulmón al atacar una señal de crecimiento defectuosa llamada EGFR. Sin embargo, para la mayoría de los pacientes estos fármacos dejan de funcionar en pocos años a medida que el cáncer desarrolla resistencia. Este estudio revela un giro sorprendente: una vez que los tumores se vuelven resistentes a los inhibidores de EGFR, desarrollan un nuevo talón de Aquiles que puede atacarse con un tipo distinto de compuesto. Comprender esta debilidad oculta podría inspirar estrategias terapéuticas futuras que acorralen la evolución del cáncer en lugar de perseguirla continuamente.
Una debilidad oculta revelada
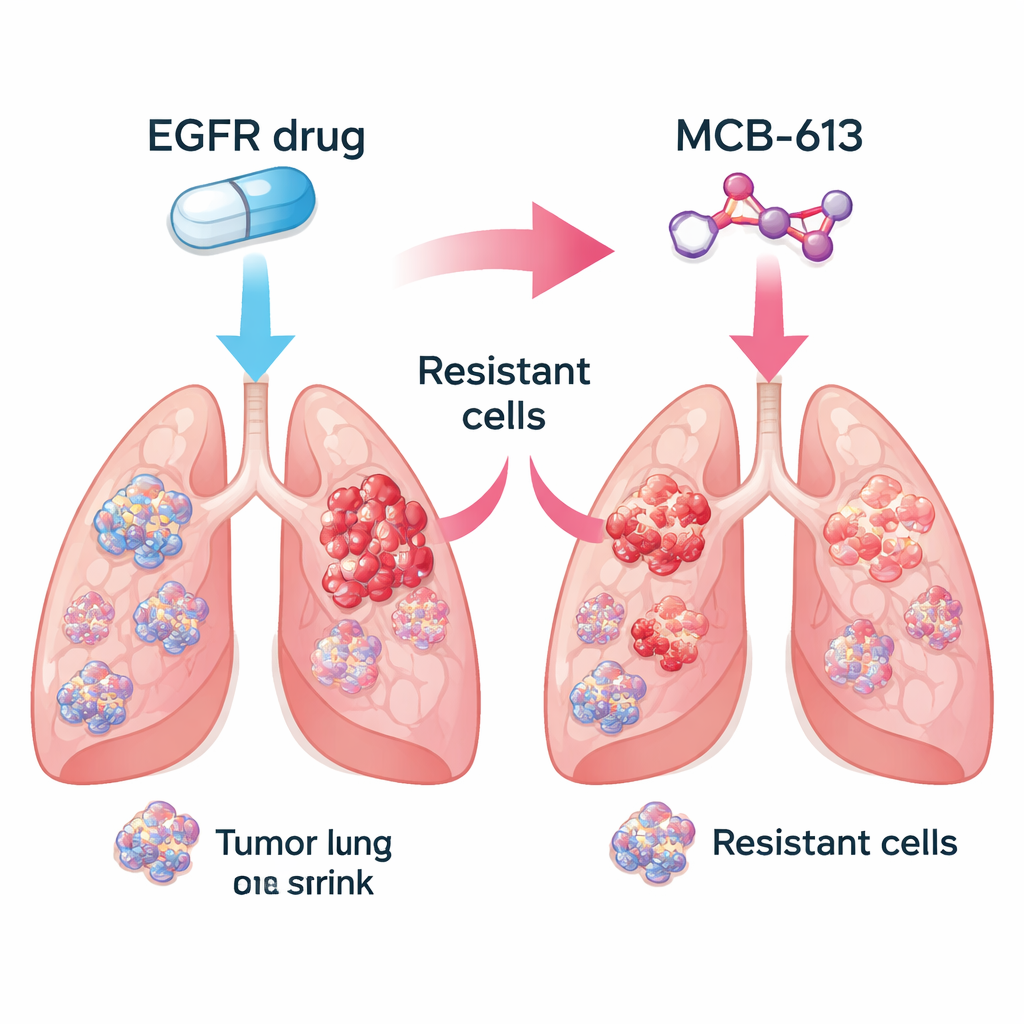
Los investigadores se centraron en cánceres de pulmón no microcíticos impulsados por EGFR mutante, una forma común de la enfermedad. En el laboratorio compararon células cancerosas sensibles a fármacos con células estrechamente relacionadas que habían evolucionado resistencia a medicamentos que bloquean EGFR como gefitinib y osimertinib. Después probaron una biblioteca de unas 2.100 pequeñas moléculas para ver cuáles mataban con más eficacia a las células resistentes que a las originales sensibles al fármaco. Entre muchos candidatos, destacó de forma consistente un compuesto llamado MCB-613. Las células resistentes que ignoraban los inhibidores de EGFR resultaron ser inusualmente vulnerables a MCB-613, tanto en placas de cultivo como en tumores de ratón.
Atrapar poblaciones tumorales mixtas Figure 1.
Los tumores reales son mezclas de células: algunas siguen siendo sensibles al fármaco original, mientras que otras adquieren resistencia mediante distintos trucos genéticos. El equipo se preguntó si combinar un inhibidor de EGFR con MCB-613 podría eliminar esta diversidad. En un experimento controlado mezclaron mayoritariamente células sensibles con una pequeña fracción de varios tipos resistentes, imitando el tumor de un paciente. Tratar esta población mixta con el inhibidor de EGFR o con MCB-613 por separado permitió que algunas células sobrevivieran y proliferaran. Pero cuando se usaron ambos agentes juntos, toda la población colapsó. Esto sugiere que emparejar una terapia dirigida estándar con un fármaco de “sensibilidad colateral” seleccionado cuidadosamente podría empujar a los tumores a un callejón evolutivo sin salida.
Un puente molecular que descompone a un guardián
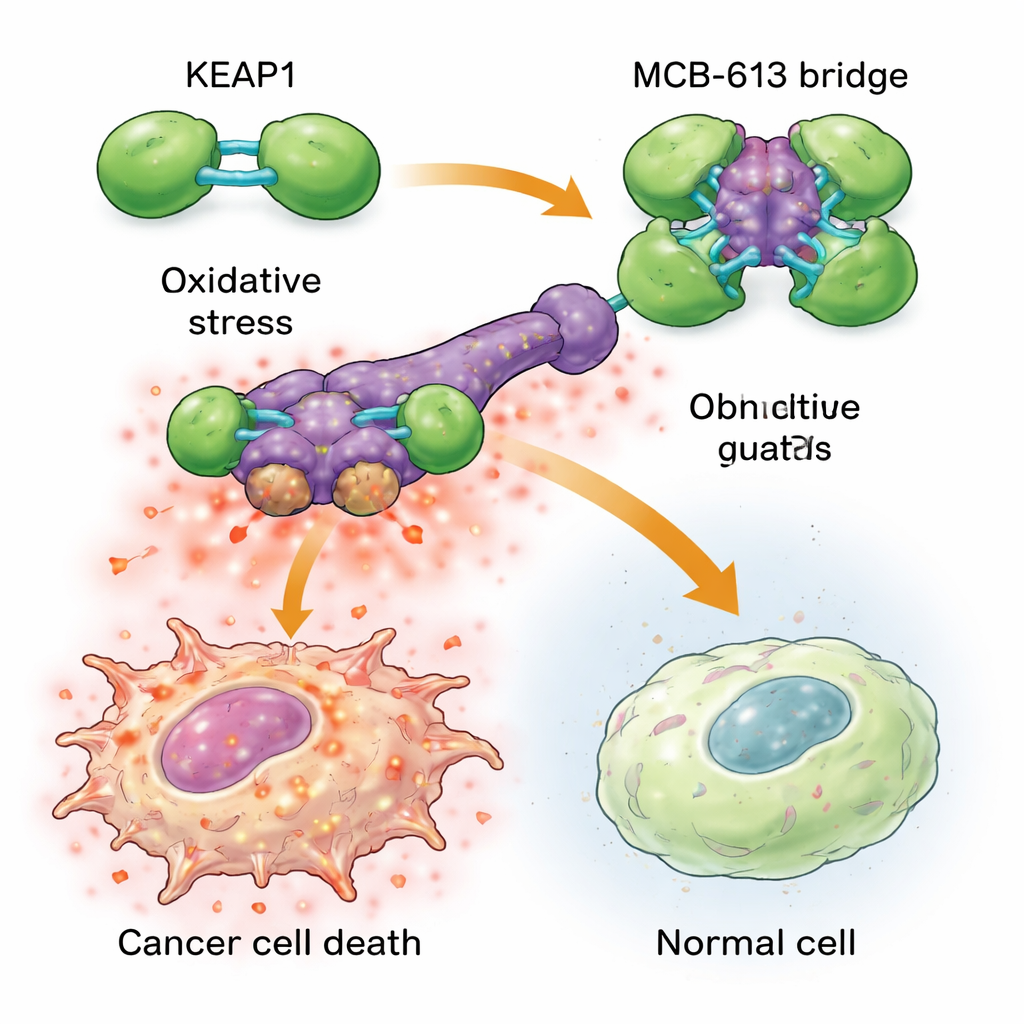
Para entender por qué MCB-613 golpea tan fuerte a las células resistentes, los científicos examinaron a qué proteínas se une. Usando sondas químicas y una pantalla dirigida de corte génico con CRISPR, identificaron a una proteína llamada KEAP1 como esencial para el efecto de MCB-613. KEAP1 actúa normalmente como un guardián celular, detectando el estrés y ayudando a regular respuestas protectoras. El equipo descubrió que MCB-613 se adhiere a KEAP1 de una manera inusual: se comporta como un puente molecular rígido que enlaza unidades de KEAP1 entre sí en agregados sobredimensionados y anormales. Este proceso no depende de los sitios reactivos de azufre habituales en KEAP1, sino de una lisina específica en su región de dimerización. Cuando esa lisina se mutó, MCB-613 ya no pudo agrupar KEAP1 y las células resistentes dejaron de ser hipersensibles al compuesto.
Convertir un estrés útil en una sobrecarga letal Figure 2.
El agrupamiento de KEAP1 desencadena una cadena de reacciones peligrosas dentro de las células cancerosas resistentes a fármacos. Estas células ya viven bajo un estrés basal más alto, con niveles elevados de especies reactivas de oxígeno (subproductos químicos dañinos) y mayor actividad en una red protectora conocida como la respuesta integrada al estrés. Cuando se añade MCB-613, la disrupción de KEAP1 lleva este estado estresado al límite: las especies reactivas de oxígeno se acumulan aún más y reguladores clave del estrés llamados ATF4 y CHOP activan programas de muerte potentes. Bloquear estos reguladores del estrés, o eliminar químicamente las especies reactivas de oxígeno, protegió en gran medida a las células frente a MCB-613. Curiosamente, el socio clásico de KEAP1, NRF2, a menudo considerado el principal impulsor de las defensas antioxidantes, no fue el responsable de la muerte; de hecho, eliminar NRF2 hizo a las células aún más sensibles, lo que subraya que MCB-613 está explotando una vía diferente y no canónica.
Qué podría significar esto para tratamientos futuros
Por ahora, MCB-613 en sí es un compuesto de laboratorio con inconvenientes químicos que lo hacen inapropiado como fármaco. Pero revela un concepto potente: a medida que los cánceres de pulmón desarrollan resistencia a los inhibidores de EGFR, pueden quedar atrapados en un estado de estrés que puede dirigirse selectivamente con compuestos que forzan a KEAP1 a formar ensamblajes disfuncionales. En principio, versiones refinadas de tales “puentes moleculares” podrían desarrollarse para ser más seguras y precisas, ofreciendo a los oncólogos una forma de conducir a los tumores hacia una “elección imposible” entre sensibilidad a la terapia dirigida original y sensibilidad al agente seguidor que induce estrés. Esta estrategia de trampa evolutiva podría, en última instancia, ayudar a retrasar o superar la resistencia en el cáncer de pulmón con mutación en EGFR y quizá en otros cánceres difíciles de tratar.
Cita: Bassil, C.F., Dillon, K., Anderson, G.R. et al. EGFR inhibitor-resistant lung cancers exhibit collateral sensitivity to a covalent, cysteine-independent KEAP1 oligomerizing molecular bridge.
Nat Commun17, 1726 (2026). https://doi.org/10.1038/s41467-026-68424-1
Palabras clave: Cáncer de pulmón con mutación en EGFR, resistencia a fármacos, sensibilidad colateral, KEAP1, estrés oxidativo