Clear Sky Science · es
Usar las referencias lineales del pangenoma para descubrir variantes de autismo perdidas
Por qué importan los cambios de ADN ocultos en el autismo
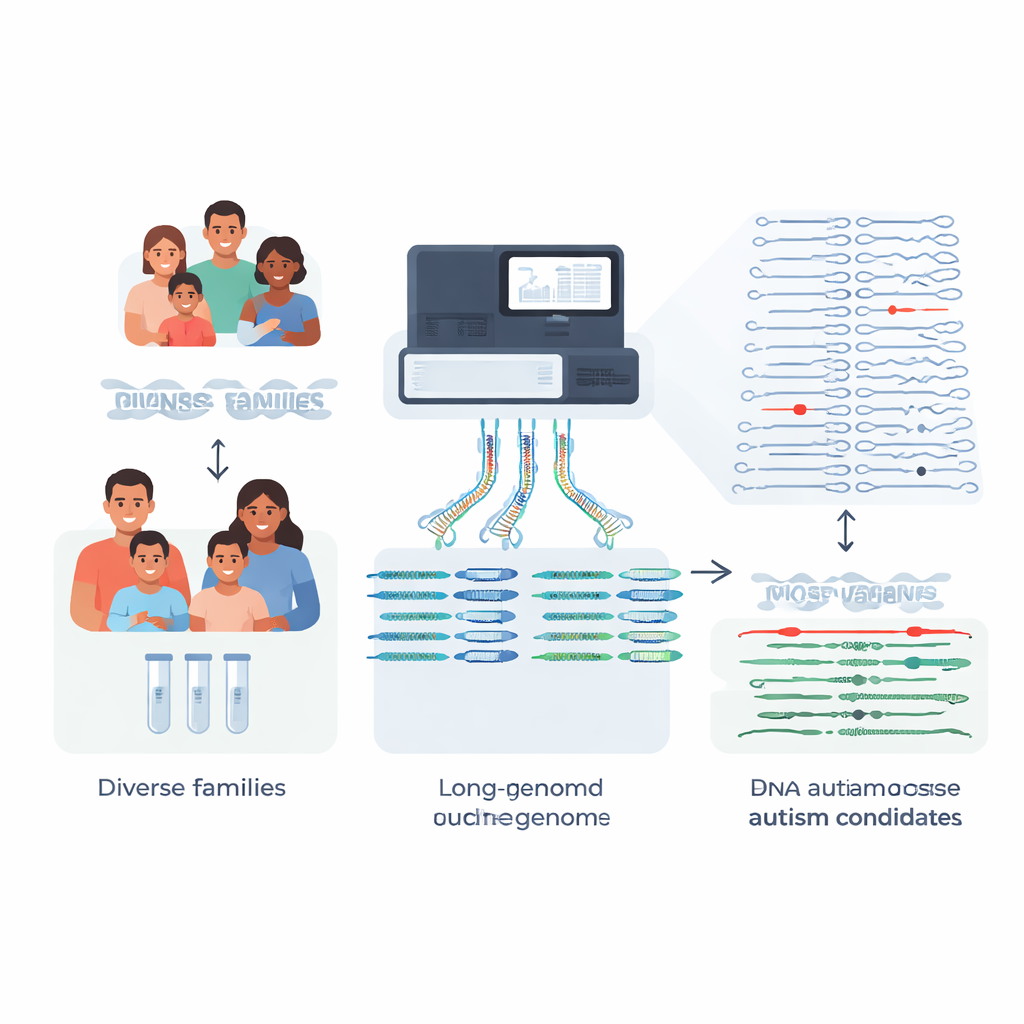
La mayoría de las familias que buscan pruebas genéticas para un niño autista esperan respuestas claras, pero aproximadamente cuatro de cada cinco no reciben una explicación genética definitiva. Este estudio aborda una razón clave: muchos cambios de gran impacto en el ADN son demasiado complejos para que las pruebas estándar los detecten. Al construir genomas casi completos de 189 personas de 51 familias afectadas por el autismo y compararlos con una nueva y más rica referencia «pangenoma», los investigadores muestran cómo la secuenciación avanzada puede descubrir mutaciones raras e invisibles hasta ahora que podrían ayudar a explicar algunos casos de autismo y afecciones relacionadas.
Más allá de las pruebas genéticas estándar
Las pruebas clínicas tradicionales se basan en fragmentos cortos de ADN para escanear el genoma de una persona. Esto funciona bien para muchos cambios de una sola letra, pero a menudo falla en regiones repetitivas o estructuralmente complejas, justo donde se esconden algunas mutaciones potentes que causan enfermedad. El equipo se centró en familias en las que pruebas previas con lecturas cortas del genoma, exoma o paneles de genes no habían encontrado la causa del autismo o de síntomas tipo Rett. Usando secuenciación de lecturas largas, que lee tramos mucho mayores de ADN, construyeron ensamblajes del genoma de alta calidad y fase para 189 individuos. Eso les permitió reconstruir las dos copias de cada cromosoma de cada persona, una heredada de cada progenitor, con muy pocas lagunas.
Variantes estructurales: cambios grandes con efectos importantes
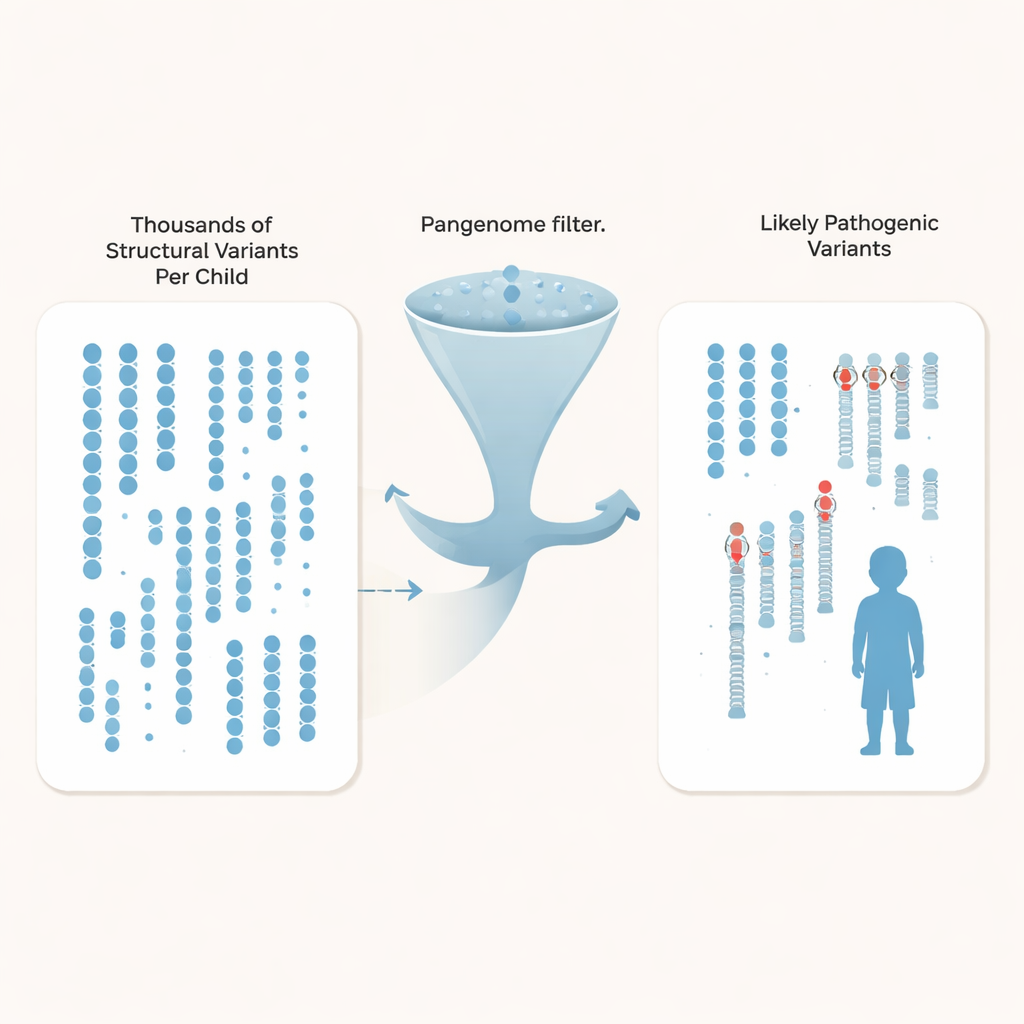
En lugar de limitarse a rastrear diferencias de una sola letra, los investigadores se centraron en variantes estructurales: inserciones, deleciones y reordenamientos que afectan al menos 50 bases de ADN y pueden alterar genes o sus interruptores regulatorios. Cada niño portaba alrededor de 27.000 de estas variantes, pero la gran mayoría son diferencias de fondo benignas compartidas en la población. Al comparar sus familias con cientos de genomas de control del pangenoma, secuenciados en profundidad y procedentes de diversas ascendencias, el equipo pudo filtrar más del 97% de las variantes estructurales comunes por cada niño, dejando aproximadamente 600 candidatos raros por genoma, y tan solo cerca de 200 cuando se usa el conjunto de control más amplio.
Encontrar mutaciones pasadas por alto en genes de riesgo conocidos
Con el espacio de búsqueda drásticamente reducido, los autores integraron varias líneas de evidencia: genes ya vinculados al autismo y a trastornos del neurodesarrollo, regiones regulatorias activas en la corteza humana en desarrollo y patrones de herencia dentro de cada familia. Descubrieron tres mutaciones claramente patógenas que las pruebas anteriores no habían detectado. Entre ellas hay una nueva señal de término en el gen SYNGAP1, importante para la función sináptica, y una deleción que elimina el último exón de MECP2, un gen clave del síndrome de Rett, aun cuando el paciente se había sometido a múltiples pruebas clínicas previas. También confirmaron un cambio causante de enfermedad en TBL1XR1, un gen que interactúa con MECP2. En total, destacaron nueve variantes estructurales adicionales —a menudo heredadas y localizadas en regiones regulatorias cercanas a genes relacionados con el cerebro— como candidatos sólidos para pruebas funcionales futuras.
Lo que el estudio no encontró —y por qué eso sigue siendo relevante
A pesar de esta búsqueda exhaustiva, los autores no observaron un exceso global claro de variantes estructurales en niños autistas frente a sus hermanos no afectados, al menos con este tamaño de muestra modesto. Hubo, sin embargo, una señal de más cambios estructurales en el cromosoma X en las niñas afectadas, y los ensamblajes casi completos de X e Y les permitieron detectar patrones inusuales como un sesgo extremo en la inactivación del cromosoma X. Estas características podrían convertirse en pistas importantes a medida que se estudien más familias. De manera crucial, el trabajo demuestra que la secuenciación de lecturas largas puede recuperar variantes patógenas que los métodos de lecturas cortas pasan por alto, sobre todo en partes difíciles del genoma y en las regiones de control que afinan la actividad génica.
Qué significa esto para las familias y las pruebas futuras
Para las familias, el impacto inmediato es modesto pero significativo: entre estos casos difíciles de resolver, alrededor del 6% recibió un diagnóstico genético claro, y casi una de cada cinco obtuvo nuevos candidatos fuertes a investigar. Para el campo, el mensaje es mayor. A medida que se incorporen más genomas de referencia completos y diversos al pangenoma y la secuenciación de lecturas largas sea más accesible, los clínicos podrán descartar cambios estructurales comunes y centrarse rápidamente en un pequeño conjunto de variantes raras y potencialmente dañinas en cada paciente. Ese cambio podría convertir gradualmente muchos de los casos de autismo “sin resolver” de hoy en otros en los que la biología subyacente —y las posibles vías para apoyo y tratamiento— se entiendan mucho mejor.
Cita: Sui, Y., Lin, J., Noyes, M.D. et al. Using the linear references from the pangenome to discover missing autism variants. Nat Commun 17, 1681 (2026). https://doi.org/10.1038/s41467-026-68378-4
Palabras clave: genética del autismo, secuenciación de lecturas largas, variantes estructurales, pangenoma humano, síndrome de Rett