Clear Sky Science · es
Diseño computacional de ciclopropanas generalistas con selectividad estereodivergente
Por qué importan las pequeñas estructuras de tres anillos en los fármacos
Los ciclopropanos —anillos de carbono de tres miembros— son bloques de construcción diminutos y tensionados que pueden cambiar drásticamente el comportamiento de un fármaco en el organismo. La disposición 3D exacta de los átomos (su estereoquímica) puede decidir si una molécula se convierte en un medicamento útil o en un análogo inactivo o incluso perjudicial. Este artículo describe una estrategia computacional para diseñar enzimas que puedan producir de forma fiable las cuatro formas 3D posibles de estos anillos a partir de los mismos materiales de partida, abriendo la puerta a una exploración más rápida y limpia de candidatos farmacéuticos.
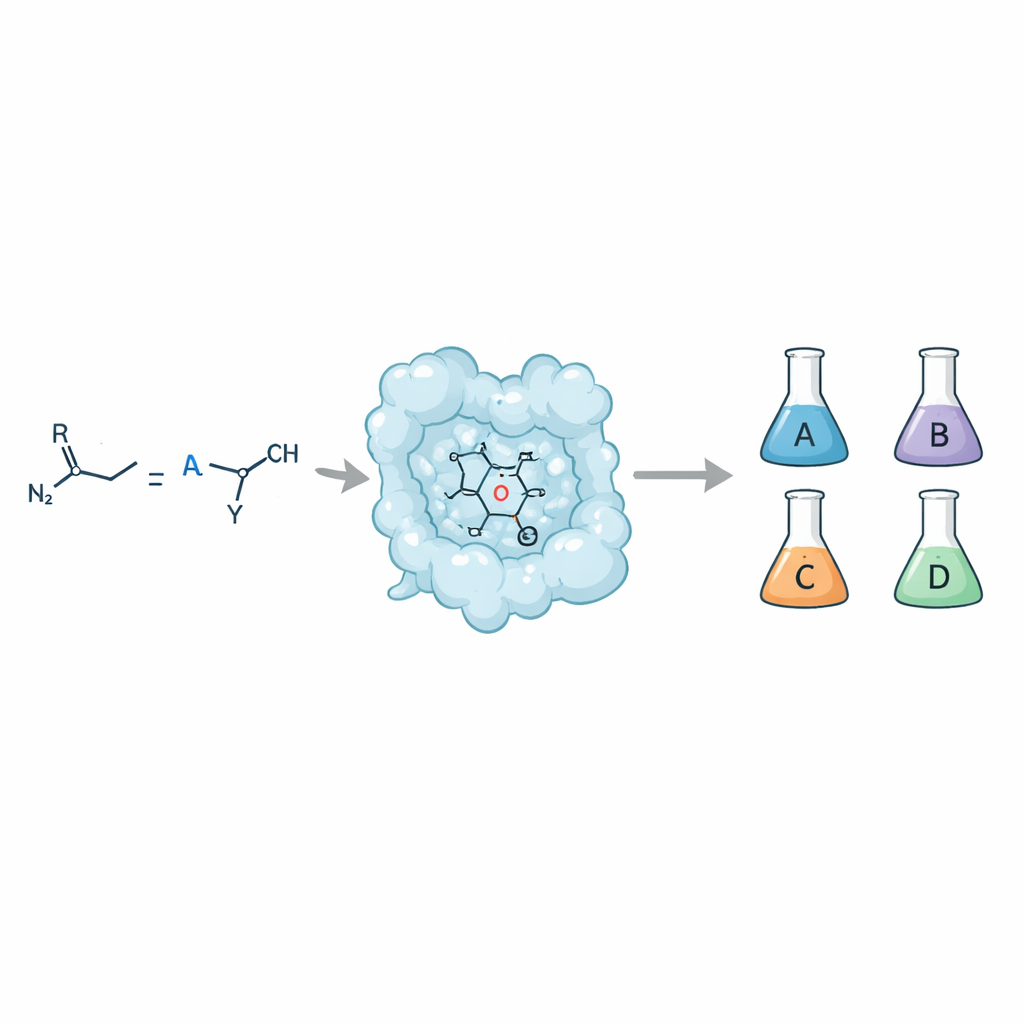
De una sola receta a cuatro resultados diferentes
Cuando un enlace doble sencillo (un olefino) reacciona con un donador de carbino como un compuesto diazo, el resultado puede ser un anillo de ciclopropano. Pero ese anillo puede adoptar cuatro formas estereoisoméricas distintas, cada una con los mismos átomos pero dispuestos de forma diferente en el espacio. Los químicos quieren acceso a cada una de estas formas porque pueden interactuar de manera muy diferente con dianas biológicas e influir en propiedades clave de los fármacos como la absorción, el metabolismo y la seguridad. Los catalizadores de pequeñas moléculas pueden a veces dar este control, pero hacerlo con enzimas —los propios catalizadores de la naturaleza— ha sido difícil, especialmente cuando se busca tanto alta selectividad como tolerancia a una amplia variedad de sustratos.
Diseñar enzimas en la pantalla del ordenador
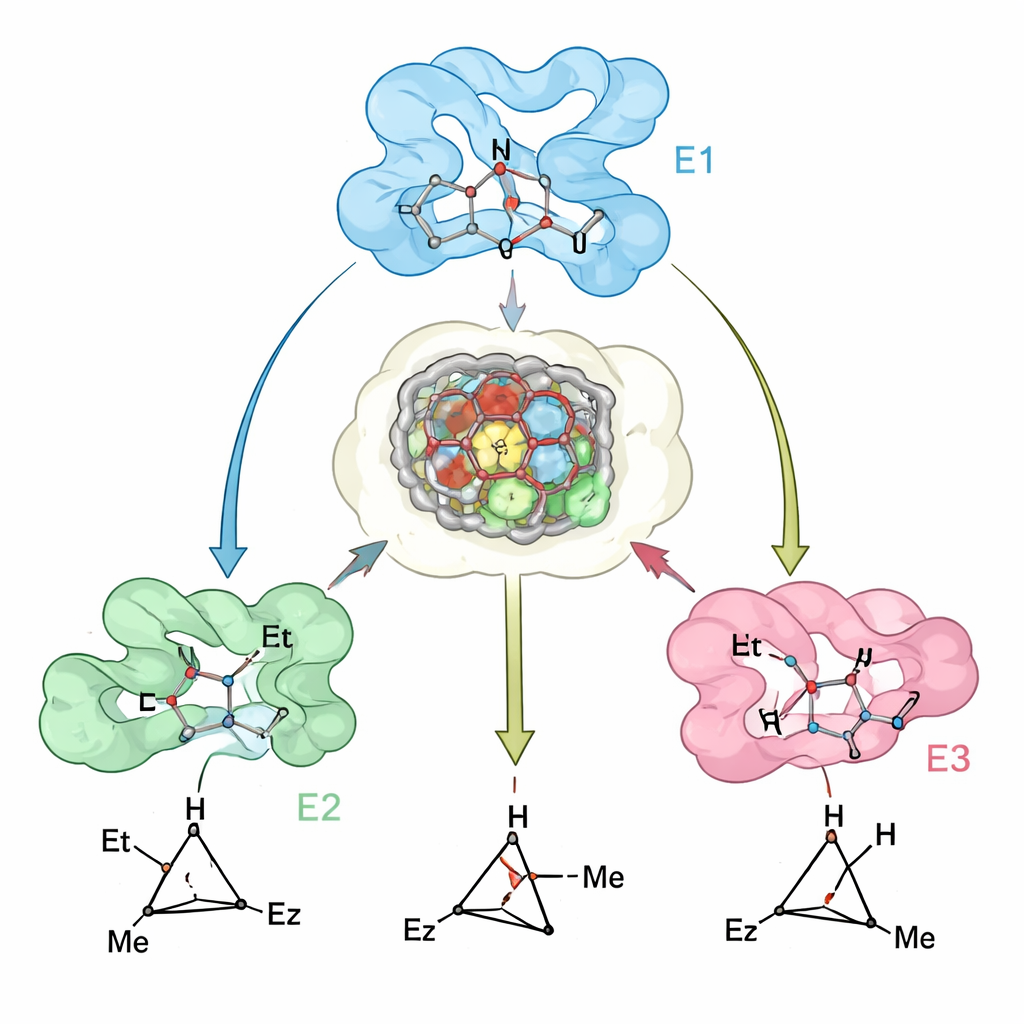
Los autores desarrollaron un flujo de trabajo computacional multietapa basado en el mecanismo para resolver este problema. Primero emplearon cálculos químicos cuánticos para modelar los efímeros estados de transición —las estructuras de alta energía a lo largo de la vía de reacción— para la formación de cada uno de los cuatro estereoisómeros de ciclopropano. Estos modelos se situaron luego en los sitios activos de diferentes proteínas que contienen hemo, y se utilizó el software de diseño de proteínas Rosetta para evaluar cuánto estabilizaba o desestabilizaba cada proteína cada estado de transición. De forma crucial, la puntuación de diseño premiaba las mutaciones que favorecían el estado de transición deseado (diseño positivo) y desfavorecían los competidores (diseño negativo), enseñando efectivamente a la enzima a “preferir” un producto 3D sobre los demás.
Construyendo una caja de herramientas enzimática completa
Con este enfoque, el equipo creó una familia de enzimas ciclopropanas “generalistas”. Partiendo de la mioglobina, rediseñaron su sitio activo para obtener variantes que producen el ciclopropano trans-(1R,2R) con muy alta selectividad y buena actividad en más de 20 olefinas diferentes, incluidos sustratos exigentes no activados y pobres en electrones. Una mioglobina previamente diseñada ya suministraba el producto complementario trans-(1S,2S). Para alcanzar los dos productos cis, los autores recurrieron a otras proteínas hemo. Remodelaron la enzima bacteriana P450cam para obtener variantes que dan selectivamente el producto cis-(1S,2R), y reorientaron la indolamina 2,3-dioxigenasa‑1 humana (IDO1) —no utilizada anteriormente para química de carbinos— para favorecer el producto cis-(1R,2S). En conjunto, estos cuatro biocatalizadores pueden entregar cada estereoisómero del mismo conjunto de productos de ciclopropano, a menudo con hasta un 99% de control tanto de diastereómero como de enantiomero.
Ver cómo el diseño coincide con la realidad
Para comprobar hasta qué punto sus modelos computacionales reflejaban enzimas reales, los investigadores resolvieron estructuras cristalinas de una variante clave de mioglobina y las compararon con las estructuras predichas. El acuerdo fue cercano, y los datos experimentales destacaron una característica sutil pero importante: el sitio activo de la proteína está preorganizado para acoger el estado de transición preferido, mientras que pequeños desplazamientos en bucles y hélices cercanos hacen que la unión del estado de transición “equivocado” sea energéticamente desfavorable. Donde las predicciones fueron menos precisas —por ejemplo, para algunos sustratos voluminosos—, las discrepancias pudieron rastrearse a movimientos del esqueleto peptídico que no fueron capturados completamente en el modelado, lo que sugiere vías claras para mejorar los métodos de diseño futuros.
Qué significa esto para futuros fármacos y catalizadores
Al combinar el modelado de estados de transición basado en física con un rediseño inteligente de proteínas, este trabajo demuestra que los resultados estereoquímicos de reacciones catalizadas por enzimas pueden programarse de antemano, en lugar de descubrirse sólo mediante evolución por ensayo y error. La suite resultante de ciclopropanas ofrece a los químicos una forma práctica de obtener juegos completos de estereoisómeros de ciclopropano a partir de una amplia gama de olefinas de partida, simplificando en gran medida los estudios de relación estructura‑actividad en descubrimiento de fármacos y síntesis de productos naturales. La misma estrategia debería poder adaptarse a otros tipos de enzimas y clases de reacciones, acelerando la creación de biocatalizadores que proporcionen un control 3D preciso sobre moléculas complejas.
Cita: Shen, Z., Siriboe, M.G., Ren, X. et al. Computational design of generalist cyclopropanases with stereodivergent selectivity. Nat Commun 17, 1620 (2026). https://doi.org/10.1038/s41467-026-68327-1
Palabras clave: biocatálisis, ciclopropanación, diseño de enzimas, estereoquímica, proteínas hemo