Clear Sky Science · es
Buenas prácticas y herramientas en R y Python para el procesamiento estadístico y la visualización de datos de lipidómica y metabolómica
Por qué importa convertir los números de laboratorio en imágenes claras
Los instrumentos modernos pueden medir ahora miles de pequeñas moléculas—lípidos y otros metabolitos—en una sola gota de sangre o tejido. Estas mediciones contienen pistas sobre riesgos de enfermedad, respuestas al tratamiento y cómo nuestro cuerpo reacciona a la dieta o al envejecimiento. Pero la salida bruta no es una respuesta lista para usar: es una enorme tabla de números que debe limpiarse, analizarse y transformarse en imágenes comprensibles. Este artículo explica cómo los investigadores pueden usar dos lenguajes de programación populares, R y Python, para hacerlo de forma fiable, transparente y con gráficos adecuados para publicación.
De mediciones químicas a tablas de datos complejas
En lipidómica y metabolómica, la espectrometría de masas y la cromatografía generan grandes conjuntos de datos donde cada fila es una muestra y cada columna es una molécula. Estas tablas rara vez se comportan como ejemplos ordenados de libro de texto. Contienen valores faltantes, valores atípicos y distribuciones sesgadas en las que unas pocas moléculas muestran niveles extremadamente altos. Las concentraciones pueden abarcar varios órdenes de magnitud y verse influidas por la edad, el sexo, la dieta, medicamentos, ritmos diarios y problemas técnicos como deriva del instrumento o efectos de lote. Grupos de expertos internacionales han emitido guías para estandarizar la recolección, el procesamiento y el reporte de muestras, pero incluso con buenas prácticas de laboratorio, un procesamiento estadístico cuidadoso sigue siendo esencial para extraer señales biológicas reales de este fondo ruidoso.
Limpieza y preparación de los números
Antes de que cualquier comparación entre grupos sanos y enfermos tenga sentido, los datos deben prepararse. La revisión describe cómo surgen los valores faltantes—por percances aleatorios, limitaciones del instrumento o interferencia de señal—y explica cuándo pueden ignorarse con seguridad, cuándo deben repetirse las mediciones y cómo pueden estimarse de forma sensata (imputarse) usando métodos como k-vecinos más cercanos, bosques aleatorios o sustituciones simples por valores bajos. A continuación, los autores exponen estrategias de normalización que reducen la variación no deseada, por ejemplo corrigiendo efectos de lote con muestras de control de calidad o ajustando por diferencias en la cantidad de muestra. Después discuten transformaciones como los logaritmos—que doman colas largas a la derecha en los datos—y métodos de escalado que ponen todas las moléculas en una base comparable para que los compuestos muy variables no dominen los análisis posteriores. 
Pruebas estadísticas y relatos visuales
Una vez que los datos están correctamente preparados, entra en juego una gama de herramientas estadísticas. Para moléculas individuales, los investigadores pueden calcular cambios en pliegue (fold changes) y usar pruebas clásicas como la t de Student o sus equivalentes no paramétricos (como la prueba de Mann–Whitney) para preguntar si los niveles difieren entre grupos. Para comparaciones que involucran varios grupos, se introducen métodos como ANOVA o la prueba de Kruskal–Wallis, acompañados de procedimientos post hoc para identificar qué grupos difieren. El poder de estas pruebas se desbloquea cuando sus resultados se visualizan con claridad. El artículo destaca diagramas de caja (incluyendo versiones mejoradas para datos sesgados), diagramas de violín y gráficos volcán que combinan tamaño del efecto y significación estadística. Para lípidos se describen visualizaciones más especializadas, como redes lipídicas que muestran cambios coordinados a través de clases completas y gráficos de cadenas aciloinsaturadas que revelan patrones en la longitud de cadena de carbono y la saturación.
Ver patrones en muchas variables a la vez
Dado que cada muestra puede tener cientos o miles de moléculas medidas, los métodos multivariantes son cruciales. La revisión explica cómo el análisis de componentes principales (PCA) comprime esta complejidad en unos pocos ejes nuevos que capturan las principales direcciones de variación, permitiendo comprobaciones rápidas de separación de grupos, efectos de lote o estabilidad analítica. Métodos no lineales más avanzados, incluidos t-SNE y UMAP, pueden revelar cúmulos y estructuras sutiles en espacios de alta dimensión. Para situaciones donde el objetivo es clasificar muestras—por ejemplo, distinguir pacientes de controles—los autores describen enfoques supervisados basados en mínimos cuadrados parciales y su extensión ortogonal (PLS-DA y OPLS-DA). Estos métodos vinculan perfiles moleculares con etiquetas de muestra, facilitan la selección de características y suelen resumirse con diagramas de scores, cargas y curvas ROC (curvas de característica operativa del receptor).
Conjuntos de herramientas prácticas en R y Python
Para ayudar a los principiantes a pasar de la teoría a la práctica, el artículo repasa un amplio ecosistema de paquetes de software. En R, colecciones como tidyverse y tidymodels simplifican la manipulación de datos y el modelado, mientras que ggplot2 y paquetes complementarios como ggpubr, ggstatsplot y tidyplots facilitan la generación de figuras aptas para publicación. Bibliotecas especializadas manejan PCA, clustering y modelos basados en PLS, y paquetes de Bioconductor dan soporte a mapas de calor complejos y gráficos interactivos. En Python, pandas proporciona el manejo de tablas, mientras que matplotlib, seaborn y plotly cubren la visualización, y scikit-learn ofrece un amplio conjunto de métodos multivariantes. A lo largo del texto, los autores enfatizan ejemplos paso a paso disponibles en un GitBook acompañante, para que los lectores puedan reproducir flujos de trabajo y adaptarlos a sus propios datos. 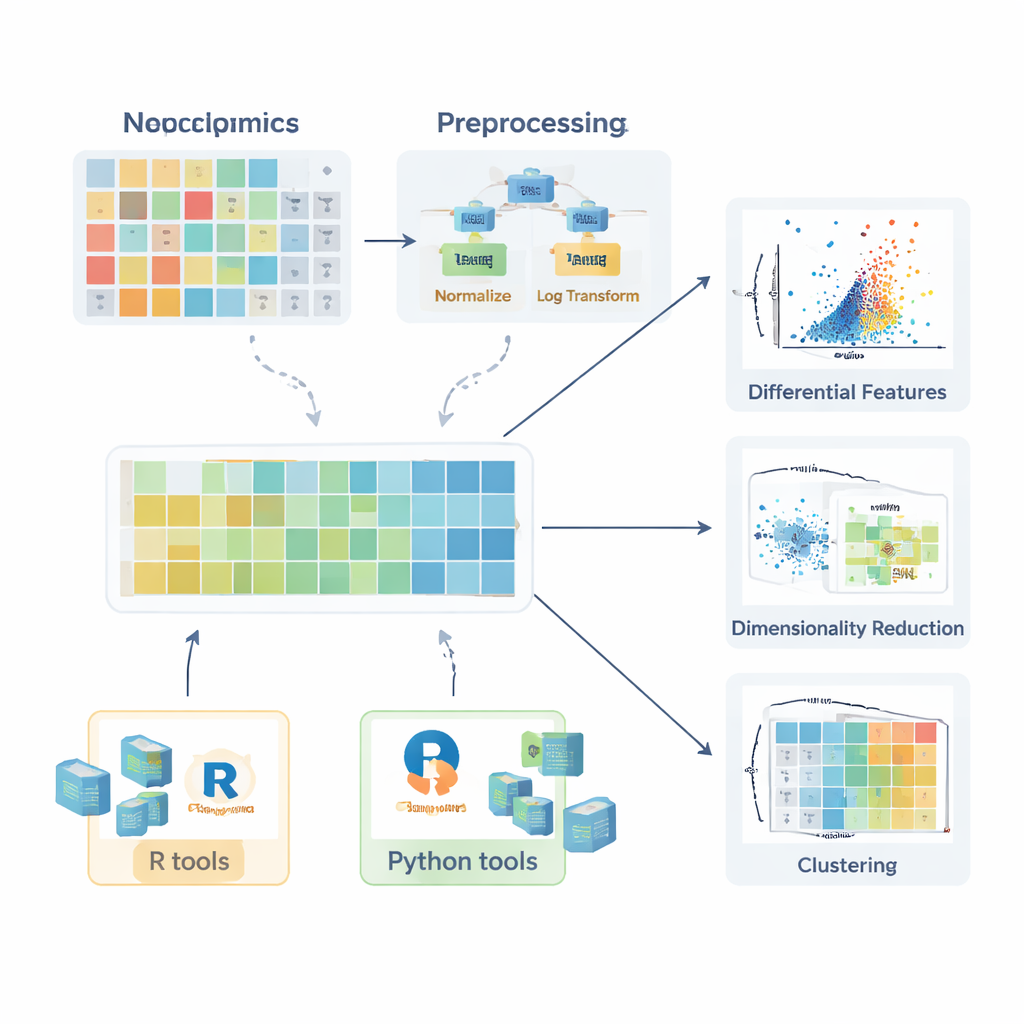
Convertir la química compleja en conocimiento fiable
El artículo concluye que la verdadera promesa de la lipidómica y la metabolómica reside no solo en instrumentos potentes, sino en la forma reflexiva en que se procesa y visualiza su salida. Siguiendo buenas prácticas estadísticas, utilizando herramientas abiertas y bien documentadas en R y Python, y apoyándose en ejemplos de código compartido, los investigadores pueden construir canalizaciones robustas y reproducibles. Esto aumenta las probabilidades de que los patrones hallados en pequeñas moléculas se traduzcan en biomarcadores fiables, un mejor entendimiento de los mecanismos de la enfermedad y enfoques más personalizados de la medicina que, en última instancia, beneficien a los pacientes.
Cita: Idkowiak, J., Dehairs, J., Schwarzerová, J. et al. Best practices and tools in R and Python for statistical processing and visualization of lipidomics and metabolomics data. Nat Commun 16, 8714 (2025). https://doi.org/10.1038/s41467-025-63751-1
Palabras clave: lipidómica, metabolómica, visualización de datos, programación R, Python