Clear Sky Science · es
Combinando transcriptómica espacial con la morfología tisular
Mirar los tejidos de dos maneras diferentes
Los médicos y los científicos cada vez quieren saber no solo qué genes están activos en un tejido, sino exactamente dónde se encienden. Al mismo tiempo, los microscopios hospitalarios ya capturan imágenes ricas de la estructura tisular que los patólogos usan a diario. Este artículo explica cómo un campo nuevo intenta conectar estas dos perspectivas —mapas detallados de actividad génica y fotografías microscópicas ordinarias— y por qué esa unión podría conducir a diagnósticos más tempranos, una mejor clasificación del cáncer y una comprensión más profunda de cómo se desarrollan y diseminan las enfermedades.
De células dispersas a mapas de actividad génica
Durante años, poderosos métodos “ómicas” exigían triturar los tejidos hasta obtener una mezcla de células individuales, lo que destruía la información sobre el origen de cada célula. La transcriptómica espacial cambió eso al medir la actividad génica conservando la posición de cada célula en el tejido. El resultado es una cuadrícula de puntos, cada uno con un perfil de expresión génica y coordenadas precisas. Por sí sola, esta información génica espacial ya ha revelado nuevos patrones de diversidad celular y arquitectura de la enfermedad. Pero no cambia una vez medida, y repetir el experimento resulta costoso. En contraste, las imágenes de tejido teñidas con colorantes estándar, como la ampliamente utilizada hematoxilina y eosina (H&E), son baratas y abundantes, y contienen pistas visuales sobre la forma celular, la densidad y la organización tisular.
Dos formas de combinar imágenes y genes
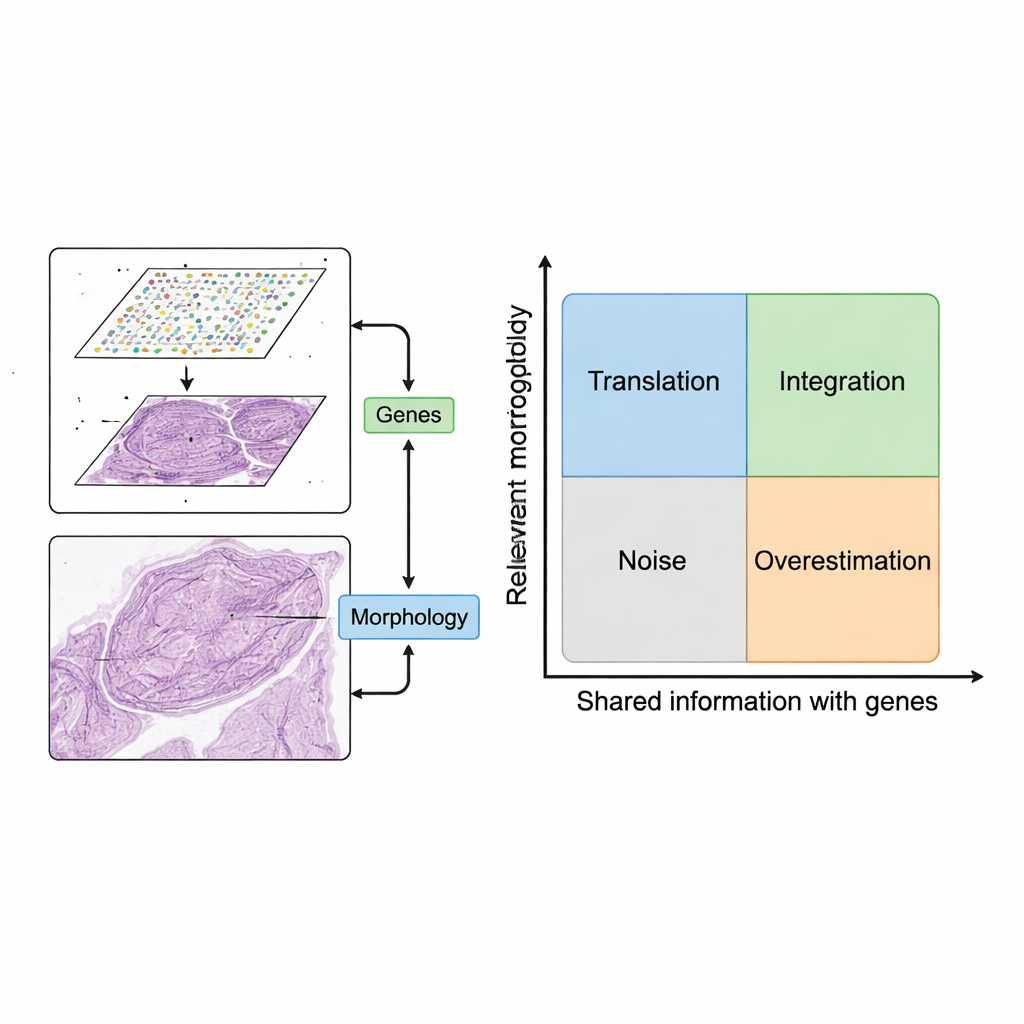
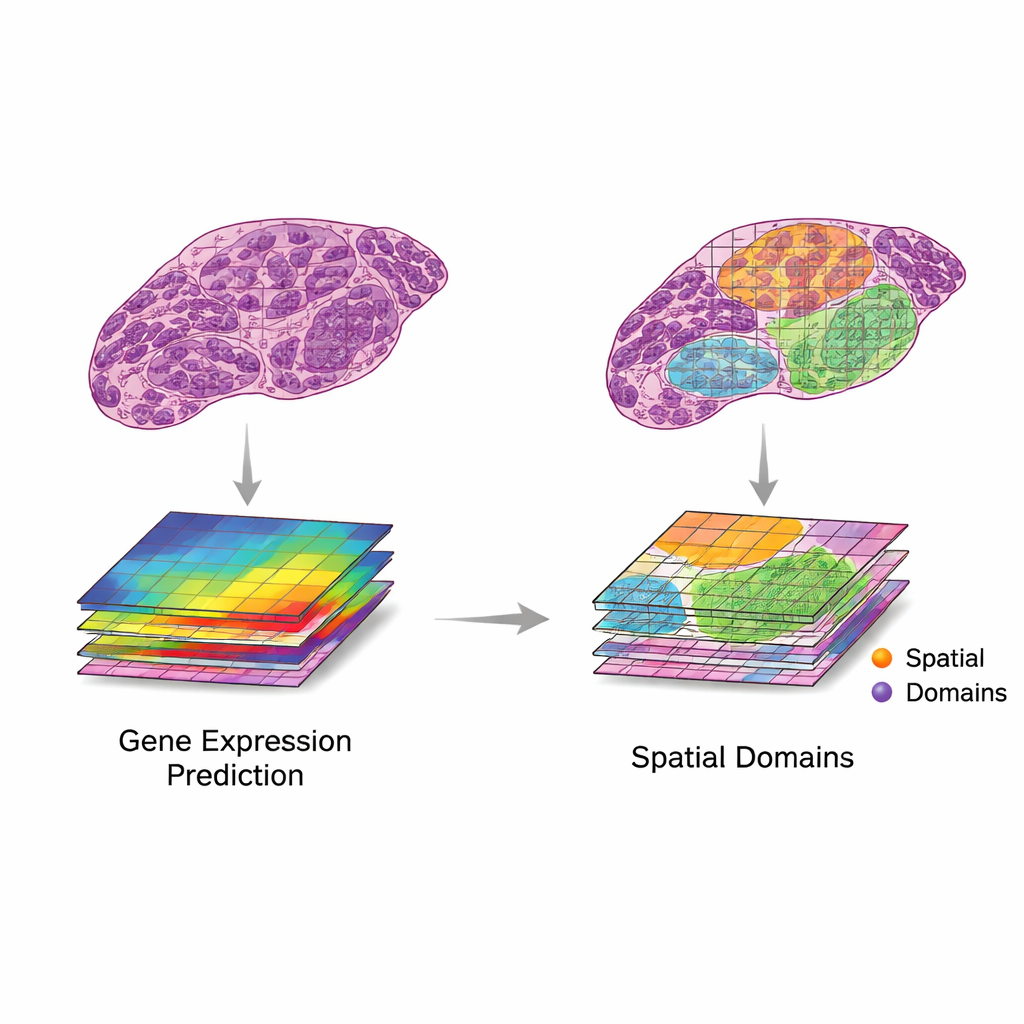
La revisión propone un marco sencillo pero potente para usar estas dos fuentes de datos en conjunto. Primero, se emparejan parches de imagen con puntos de expresión génica cercanos. Luego, modelos informáticos extraen características de las imágenes —patrones que capturan forma, textura y organización— y las comparan con patrones en la expresión génica. Los autores describen dos escenarios deseables. En la “traducción”, las características de la imagen reflejan estrechamente la actividad génica relevante, lo que permite a los modelos predecir qué genes están activos usando solo la imagen del tejido. Esto puede emplearse para completar mediciones génicas faltantes, alcanzar una resolución más fina que la cuadrícula original, o inferir actividad génica a partir de preparaciones clínicas rutinarias sin trabajo de laboratorio adicional. En la “integración”, las características de la imagen capturan información útil que los datos génicos pasan por alto, como cambios estructurales lentos o una organización tisular sutil, ayudando a definir regiones o “dominios” más claros dentro de un tejido.
Cuando la información extra ayuda —y cuando estorba
No todas las características de la imagen merecen ser usadas. Los autores introducen un mapa conceptual con dos ejes: cuán relevante es una característica de imagen para la pregunta biológica y cuánto se solapa con la información génica. Las características que no son ni relevantes ni están relacionadas con los genes equivalen a ruido, como artefactos de tinción. Las características que siguen patrones génicos pero están ligadas a genes poco importantes (como genes de mantenimiento básicos) pueden hacer que los modelos parezcan buenos en papel sin aportar valor clínico. Al organizar los métodos en cuatro cuadrantes —traducción, integración, ruido y sobreestimación— el marco aclara cuándo combinar imágenes y genes añade realmente información y cuándo simplemente repite u oculta lo que ya se conoce.
Herramientas actuales, pruebas y dificultades crecientes
Una ola rápida de métodos de inteligencia artificial intenta ahora realizar traducción e integración sobre datos reales. Los primeros sistemas se basaban en redes neuronales convolucionales, mientras que los más recientes usan transformers, redes neuronales de grafos y modelos multinivel que pueden captar desde detalles de pequeñas estructuras celulares hasta el contexto de una laminilla completa. Estos métodos se han utilizado para predecir actividad génica a partir de imágenes H&E, generar mapas de superresolución y ayudar a identificar regiones tisulares con comportamiento distinto. Para evaluar el rendimiento, los investigadores recurren a medidas estadísticas como la correlación entre niveles génicos predichos y observados, o la concordancia entre regiones definidas por la IA y las etiquetas de patólogos expertos. Sin embargo, los conjuntos de datos siguen siendo pequeños y heterogéneos, y la comparación entre estudios es difícil. Muchas mejoras reportadas pueden reflejar sobreajuste o éxito en genes y patrones que importan poco en la clínica.
Hacia dónde podría conducir esto
Los autores concluyen que combinar mapas génicos espaciales con imágenes tisulares es una iniciativa prometedora pero aún en fases tempranas. Los modelos actuales a menudo alcanzan solo una precisión moderada y todavía no están demostrados como listos para uso médico rutinario. El progreso futuro probablemente provendrá de mejores características de imagen, en especial grandes “modelos base” entrenados con millones de laminillas de patología, y de centrarse en genes y patrones que realmente influyan en la atención al paciente. Una integración diseñada con cuidado podría algún día revelar señales tempranas de enfermedad al detectar discrepancias entre cómo se ve el tejido ahora y lo que sus genes predicen que ocurrirá después. En resumen, este trabajo traza una hoja de ruta para convertir imágenes microscópicas de rutina en mapas enriquecidos por la información génica que ayuden a los médicos a entender y tratar la enfermedad con mayor precisión.
Cita: Chelebian, E., Avenel, C. & Wählby, C. Combining spatial transcriptomics with tissue morphology. Nat Commun 16, 4452 (2025). https://doi.org/10.1038/s41467-025-58989-8
Palabras clave: transcriptómica espacial, morfología tisular, patología digital, predicción de expresión génica, IA en imágenes