Clear Sky Science · es
ATGL sensibiliza las células de carcinoma hepatocelular a fármacos genotóxicos al modular el estado de acetilación/fosforilación de p53
Convertir la degradación de grasa en una vulnerabilidad del cáncer
La quimioterapia estándar para el cáncer de hígado a menudo fracasa porque las células tumorales son extraordinariamente buenas sobreviviendo al daño del ADN. Este estudio explora un aliado inesperado dentro de esas mismas células cancerosas: una enzima que descompone la grasa almacenada. Al aumentar esta enzima, llamada ATGL, los investigadores observaron que podían empujar a las células tumorales hepáticas a dejar de reparar sus lesiones en el ADN y, en su lugar, autodestruirse. El trabajo descubre un vínculo oculto entre cómo las células cancerosas manejan las grasas y cómo responden a potentes fármacos que dañan el ADN, lo que sugiere nuevas formas de mejorar la eficacia de los tratamientos existentes.
Por qué los tumores hepáticos resisten fármacos agresivos
El cáncer de hígado, en particular el carcinoma hepatocelular, es uno de los tipos tumorales más comunes y mortales a nivel mundial. Muchos pacientes reciben fármacos que dañan el ADN, como etopósido y doxorubicina, con la esperanza de forzar a las células cancerosas a una crisis letal. Sin embargo, estas células a menudo escapan deteniendo su crecimiento y activando sistemas de reparación controlados por una proteína guardiana conocida como p53. Si el daño puede repararse, las células reanudan la división; si no, p53 también puede desencadenar la muerte celular programada. El rompecabezas central es qué inclina a p53 hacia la reparación frente a la autodestrucción, y por qué algunos tumores siguen siendo tan tercamente resistentes a la terapia.
Una enzima que corta la grasa inclina la balanza
El equipo se centró en ATGL, una enzima que recorta las grasas almacenadas en pequeños depósitos celulares llamados gotas lipídicas. En los cánceres de hígado, los niveles de ATGL suelen ser inferiores a los del tejido sano, y trabajos previos sugerían que forzar a las células a producir más ATGL frenaba el crecimiento tumoral. Aquí, los investigadores diseñaron líneas celulares de cáncer hepático que sobreproducían ATGL o la reducían, y luego las expusieron a fármacos genotóxicos. Las células con ATGL extra acumularon muchas más señales de ADN roto, mientras que las con ATGL reducida mostraron menos daño. Bloquear la actividad cortante de ATGL con un inhibidor específico, o expresar una forma mutante inactiva, eliminó esta sensibilidad aumentada, demostrando que la actividad lipolítica de la enzima es clave.
Reprogramar la decisión celular: reparar o morir
Profundizando, los científicos examinaron a p53, que actúa como un agente de tráfico molecular tras una lesión en el ADN. El comportamiento de p53 se dirige mediante pequeñas etiquetas químicas añadidas en posiciones concretas. En las células ricas en ATGL, los fármacos genotóxicos hicieron que p53 ganara más de un tipo de etiqueta (grupos acetilo) y comparativamente menos de otro (grupos fosfato). Este cambio favoreció la activación de genes que promueven la muerte celular, como Puma, mientras atenuaba genes como p21 y GADD45 que normalmente detienen el ciclo celular y respaldan la reparación del ADN. Como resultado, incluso después de lavar el fármaco, las células con mucho ATGL no lograron eliminar los marcadores de daño del ADN y avanzaron hacia la apoptosis en lugar de recuperarse.
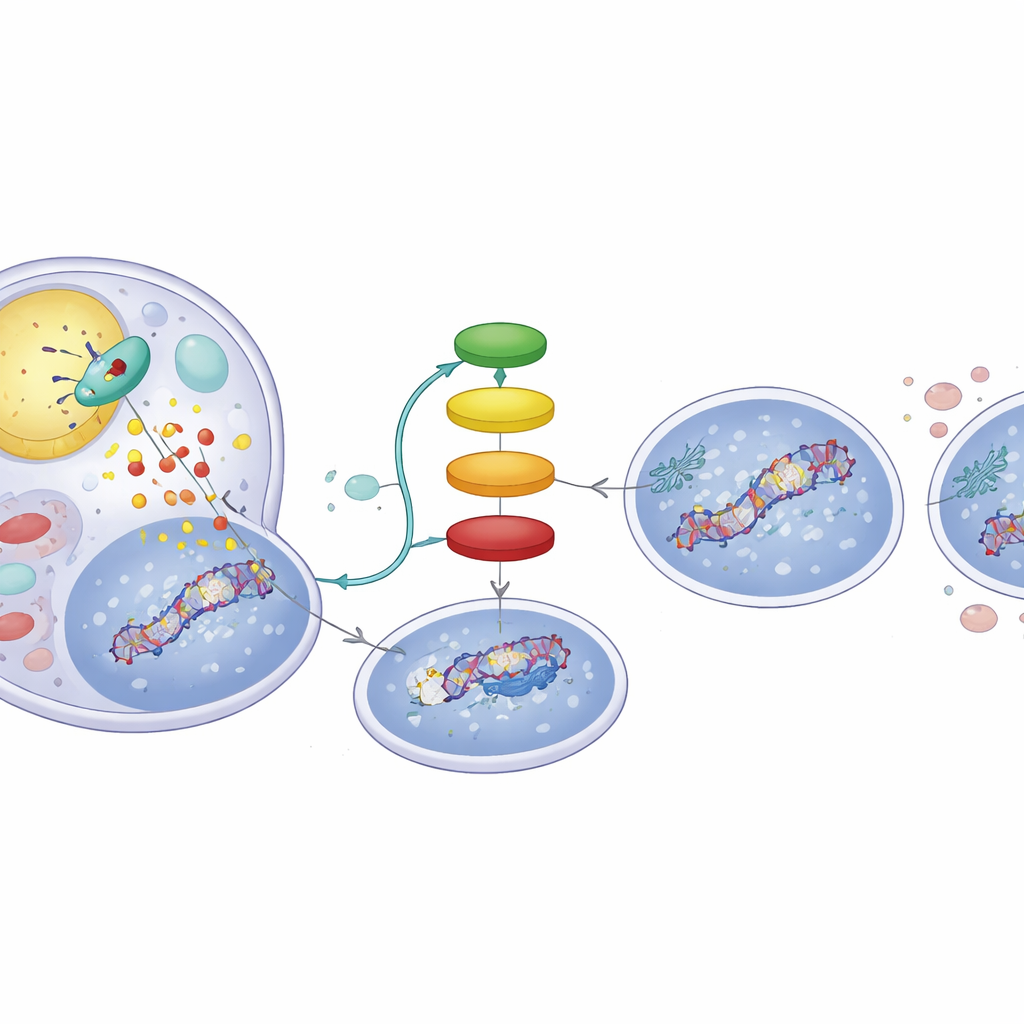
Una cadena de señales impulsada por la grasa dentro de las células tumorales
¿Cómo cambia el corte de grasas las etiquetas de p53? Los productos de la degradación por ATGL son ácidos grasos libres que pueden actuar como mensajeros. El estudio muestra que estos ácidos grasos activan un receptor nuclear llamado PPARα, que a su vez aumenta la actividad de p300, una proteína que coloca etiquetas acetilo en p53. Cuando los investigadores usaron un compuesto activador de PPARα, reprodujeron el comportamiento de alta ATGL: aumento de las señales de daño del ADN y un perfil de p53 sesgado hacia la apoptosis. A la inversa, bloquear p300 anuló los cambios inducidos por ATGL en p53 y redujo el daño del ADN, subrayando que la cadena ATGL → PPARα → p300 es central para este cambio. Los análisis de cientos de tumores hepáticos humanos en conjuntos de datos públicos avalaron este vínculo, revelando que los tumores con mayor expresión de ATGL también tienden a mostrar firmas más fuertes de PPARα y p300 y expresión de genes controlados por p53.
Qué podría significar esto para tratamientos futuros
En términos sencillos, el estudio revela que cuando se estimula a las células de cáncer de hígado a quemar la grasa almacenada mediante ATGL, son menos proclives a reparar el daño del ADN inducido por la quimioterapia y más propensas a someterse a la muerte celular ordenada. Esto sugiere dos posibilidades prácticas: medir los niveles de ATGL podría ayudar a predecir qué pacientes responderán mejor a fármacos genotóxicos, y potenciar la actividad de ATGL o su vía descendente PPARα podría emplearse junto con las quimioterapias existentes para superar la resistencia. Aunque se necesitan más pruebas en animales y pacientes, el trabajo subraya un mensaje llamativo: en el cáncer de hígado, hacer que las células tumorales sean «más delgadas» a nivel microscópico también puede hacerlas más vulnerables a fármacos que salvan vidas.
Cita: Castelli, S., De Cristofaro, A., Desideri, E. et al. ATGL sensitizes hepatocellular carcinoma cells to genotoxic drugs by modulating p53 acetylation/phosphorylation status. Cell Death Discov. 12, 164 (2026). https://doi.org/10.1038/s41420-026-03048-4
Palabras clave: carcinoma hepatocelular, ATGL, respuesta al daño del ADN, señalización de p53, metabolismo lipídico en el cáncer