Clear Sky Science · es
El estado de metilación de la fosfatasa 2A afecta la alfa-sinucleinopatía en modelos murinos
Por qué esto importa para la salud cerebral
La enfermedad de Parkinson y trastornos relacionados privan lentamente a las personas de la movilidad, la memoria y la autonomía. Un culpable principal es una proteína cerebral llamada alfa-sinucleína que puede plegarse mal, agregarse y dañar las neuronas. Este estudio plantea una pregunta esperanzadora: en lugar de atacar la proteína directamente, ¿podemos ajustar la propia maquinaria de limpieza del cerebro para evitar que la alfa-sinucleína se vuelva tóxica?
La historia de una proteína pegajosa
En la enfermedad de Parkinson y la demencia con cuerpos de Lewy, ovillos retorcidos de alfa-sinucleína se acumulan dentro de las neuronas, formando los clásicos “cuerpos de Lewy”. Una etiqueta química particular en esta proteína, añadida en un sitio llamado serina 129, está fuertemente vinculada a su forma más dañina. Cuando esta marca es abundante, la alfa-sinucleína tiende a formar fibrillas rígidas y agregados. El cerebro normalmente equilibra estas etiquetas mediante enzimas que las añaden y otras que las quitan. Como muchas enzimas pueden añadir la etiqueta, bloquear solo una de ellas probablemente no funcione. En lugar de ello, los autores se centraron en la familia principal de enzimas que elimina la etiqueta, llamada fosfatasa de proteínas 2A, o PP2A, que actúa como un borrador molecular para esta modificación peligrosa.
El borrador del cerebro y sus dos interruptores
PP2A no funciona a plena potencia por defecto. Su actividad depende de una pequeña marca química, llamada metilación, en una de sus subunidades. Dos proteínas controlan este interruptor: LCMT-1 añade la marca y orienta a PP2A hacia una forma más activa y protectora, mientras que PME-1 la elimina y empuja a PP2A hacia un estado menos activo y dañino. Trabajos previos en tejido cerebral humano mostraron que en Parkinson y en la demencia con cuerpos de Lewy, LCMT-1 tiende a ser menor y PME-1 mayor, dejando a PP2A debilitada. El estudio actual prueba directamente qué ocurre cuando estos interruptores se manipulan deliberadamente en una u otra dirección en ratones vivos que desarrollan problemas de alfa-sinucleína.
Probar el equilibrio en cerebros vivos
Los investigadores usaron dos modelos murinos complementarios. En uno, los ratones fueron diseñados para producir alfa-sinucleína humana en todo el cerebro, desarrollando gradualmente agregados proteicos y problemas de movimiento y memoria con la edad. Estos animales se modificaron además para sobreproducir PME-1 (el interruptor “apagado” de PP2A) o LCMT-1 (el interruptor “encendido” de PP2A) en neuronas del prosencéfalo. En el segundo modelo, el equipo inyectó fibrillas preformadas de alfa-sinucleína en el cuerpo estriado, una región cerebral profunda implicada en el movimiento. Estas fibrillas actúan como semillas, reclutando alfa-sinucleína normal y propagando la patología durante meses en ratones por lo demás normales o con alteraciones en las enzimas. En ambos modelos, los científicos midieron la acumulación de proteína, la salud neuronal, la inflamación cerebral y el comportamiento.
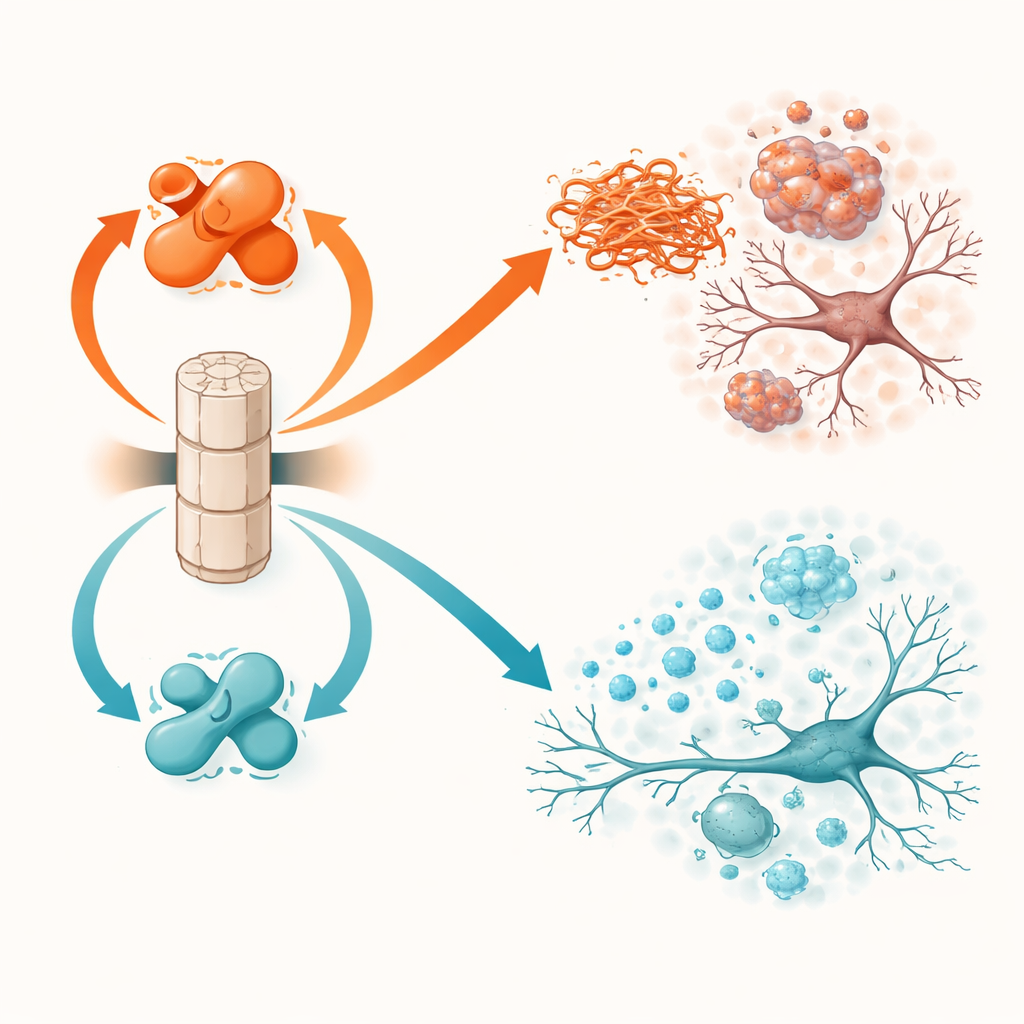
Cuando el borrador falla, el daño se extiende
Los ratones que sobreprodujeron PME-1, y por tanto tenían PP2A menos activa, evolucionaron peor. En los animales transgénicos de alfa-sinucleína, aumentar PME-1 condujo a una alfa-sinucleína más fuertemente etiquetada y agregada en corteza e hipocampo, mayor pérdida de la estructura neuronal, señales de actividad neuronal más débiles y una activación más intensa de las células inmunes del cerebro. Estos cambios se tradujeron en peores resultados en pruebas de movimiento y en tareas de aprendizaje y memoria. En el modelo de inyección de fibrillas, la sobreexpresión de PME-1 permitió que se acumulasen y propagasen más extensamente las asambleas tóxicas de alfa-sinucleína, especialmente hacia las neuronas productoras de dopamina de la sustancia negra, una región clave perdida en la enfermedad de Parkinson. Estos ratones mostraron mayor pérdida de fibras dopaminérgicas, inflamación más intensa y déficits motores y de anidamiento más acusados.
Volver a encender el borrador
La manipulación opuesta, sobreproducir LCMT-1 para mantener a PP2A fuertemente metilada y activa, tuvo efectos ampliamente protectores. En ratones transgénicos de alfa-sinucleína, LCMT-1 redujo la carga de proteína etiquetada y agregada hasta niveles cercanos a lo normal y preservó tanto la estructura como la actividad de las neuronas. Los marcadores de inflamación fueron menores y los animales se desempeñaron más cerca de los controles sanos en pruebas de equilibrio y memoria. En el modelo de siembra por fibrillas, LCMT-1 limitó tanto la acumulación local como la propagación a largo alcance de la alfa-sinucleína tóxica, protegió a las neuronas dopaminérgicas de la degeneración, redujo la activación de la microglía y atenuó el declive en la coordinación motora y el comportamiento de anidamiento. En todos los experimentos, desplazar a PP2A hacia su estado activo y metilado tradujo de forma consistente beneficios moleculares en protección funcional.
Qué podría significar esto para tratamientos futuros
Para no especialistas, la lección es directa: el cerebro tiene un borrador incorporado que puede quitar una etiqueta dañina de la alfa-sinucleína y evitar que se convierta en agregados peligrosos. Cuando este borrador se debilita, el daño, la inflamación y los síntomas empeoran; cuando se fortalece, las neuronas quedan protegidas. El estudio proporciona evidencia directa en animales vivos de que el estado de metilación de PP2A es un control maestro de la toxicidad de la alfa-sinucleína y sus consecuencias. Esto apunta a una nueva estrategia terapéutica: en lugar de perseguir cada forma nociva de la proteína, podrían diseñarse fármacos que impulsen a PP2A y a sus reguladores LCMT-1 y PME-1 hacia un estado más protector. Tales enfoques requerirán pruebas de seguridad cuidadosas, pero tienen potencial para ralentizar o prevenir la enfermedad de Parkinson y afecciones relacionadas restaurando la capacidad del cerebro para mantener la alfa-sinucleína bajo control.
Cita: Maddila, S., Hassanzadeh, K., Liu, J. et al. Protein phosphatase 2A methylation state impacts α-synucleinopathy in mouse models. Cell Death Discov. 12, 172 (2026). https://doi.org/10.1038/s41420-026-03045-7
Palabras clave: Enfermedad de Parkinson, alfa-sinucleína, fosfatasa de proteínas 2A, neurodegeneración, inflamación cerebral