Clear Sky Science · es
Modulación de firmas metabólicas para mitigar la resistencia a cabozantinib en modelos celulares de leucemia mieloide aguda con FLT3-ITD
Por qué esto importa para el tratamiento del cáncer
Muchos fármacos oncológicos modernos están diseñados para apuntar a una única proteína defectuosa en las células tumorales. Estos medicamentos dirigidos pueden provocar remisiones notables, pero los cánceres con frecuencia encuentran formas de adaptarse y reaparecer. Este artículo examina cómo un tipo de cáncer de la sangre, la leucemia mieloide aguda (LMA), desarrolla resistencia a uno de esos fármacos dirigidos, el cabozantinib, y cómo redirigir el uso energético de las células cancerosas podría ayudar a los médicos a superar esa resistencia.
Células leucémicas que aprenden a eludir un fármaco dirigido
Los investigadores se centraron en células de LMA que portan una mutación en un interruptor de señal de crecimiento llamado FLT3-ITD, conocida por impulsar una enfermedad especialmente agresiva. El cabozantinib, una píldora ya utilizada en varios tumores sólidos, puede bloquear con fuerza a las células leucémicas impulsadas por FLT3 en el laboratorio. Para modelar lo que ocurre en los pacientes con el tiempo, el equipo expuso gradualmente dos líneas celulares de LMA con mutación FLT3 a dosis crecientes de cabozantinib hasta que algunas células sobrevivieron y volvieron a proliferar. Estas nuevas poblaciones celulares, denominadas Molm13-XR y MV4-11-XR, pudieron tolerar concentraciones de cabozantinib mucho más altas que sus células “parentales” originales. También se volvieron menos sensibles a otros dos fármacos aprobados que apuntan a FLT3, sorafenib y quizartinib, mientras que seguían siendo vulnerables a un inhibidor diferente, gilteritinib.
Ajustes genéticos que ayudan a la supervivencia del cáncer
Al echar un vistazo en profundidad, los científicos hallaron que estas células leucémicas resistentes llevaban nuevos cambios en su gen FLT3. Ambas líneas resistentes habían adquirido la misma mutación puntual, llamada D835Y, en una región crucial del dominio quinasa de FLT3, un punto caliente conocido para la resistencia a varios fármacos. Una de las líneas, MV4-11-XR, también sufrió una inusual deleción de 1,3 kilobases que eliminó un exón completo de FLT3, borrando parte del dominio importante para la unión del fármaco. Estos cambios parecieron haberse seleccionado durante la exposición prolongada a cabozantinib: las versiones mutantes de FLT3 se hicieron mucho más comunes en las células resistentes que en la población inicial. Al mismo tiempo, vías de señalización clave aguas abajo de FLT3 —como ERK, STAT5 y AKT— estaban más activadas, favoreciendo un crecimiento más rápido y una mayor formación de colonias en las células resistentes.
Las células cancerosas cambian su sistema de combustible
El equipo preguntó entonces si la resistencia estaba ligada no solo a la genética, sino también a cómo se abastecían de energía las células. Mediante secuenciación de ARN y pruebas metabólicas específicas, encontraron un patrón consistente: las células resistentes a cabozantinib dependían mucho más de la glucólisis —la rápida descomposición de la glucosa en el citoplasma— incluso cuando el oxígeno era abundante. Estas células absorbían más glucosa, producían más lactato, mostraban mayor actividad de una enzima clave llamada GAPDH y aumentaban la expresión de varios genes relacionados con la glucólisis. En contraste, las mitocondrias de las células, las estructuras que sostienen una producción de energía más eficiente, estaban menos activas y eran menos abundantes. Las mediciones del uso de oxígeno revelaron que tanto la respiración mitocondrial basal como la máxima estaban reducidas, y las especies reactivas de oxígeno dentro de las células estaban elevadas, lo que apunta a mitocondrias estresadas y con bajo rendimiento.
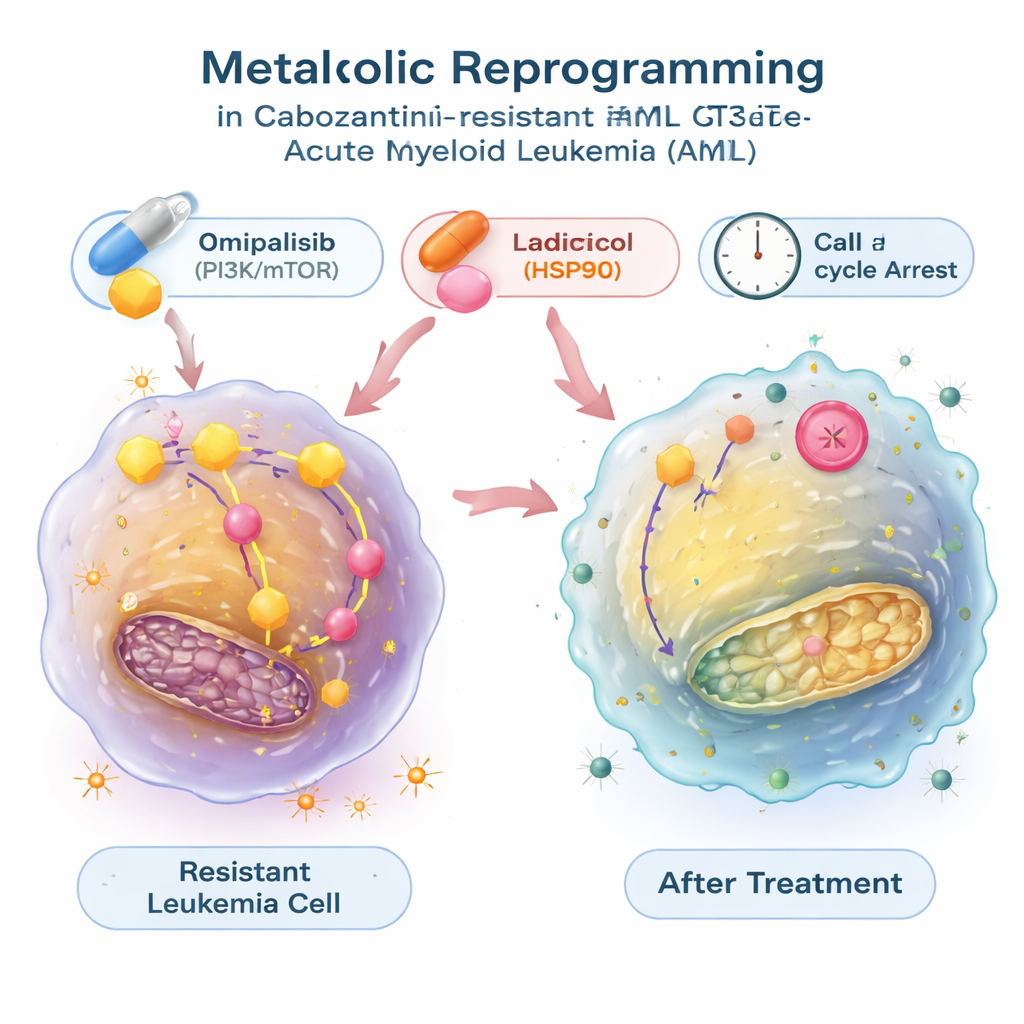
Encontrar fármacos que devuelvan el interruptor metabólico
Para ver si este cambio energético podía revertirse, los investigadores utilizaron una gran base de datos pública que relaciona patrones de expresión génica con los efectos de miles de compuestos. Buscaron fármacos predichos para contrarrestar la firma metabólica de las células leucémicas resistentes y se centraron en dos: radicicol, que bloquea una chaperona proteica llamada HSP90, y omipalisib, que inhibe la vía de señalización PI3K/mTOR que controla el crecimiento y el metabolismo. En pruebas de laboratorio, ambas moléculas no solo ralentizaron el crecimiento de las células resistentes, sino que también redujeron su glucólisis hiperactiva, normalizando la captación de glucosa y la liberación de lactato y disminuyendo la expresión de genes relacionados con la glucólisis. Estos compuestos empujaron a las células leucémicas hacia una fase de reposo del ciclo celular y, en el caso del radicicol, también desencadenaron una muerte celular programada sustancial. Cuando se combinaron con cabozantinib, omipalisib —y, en un modelo, radicicol— actuaron de forma sinérgica, facilitando la eliminación de las células resistentes al fármaco.
Qué significa esto para terapias futuras de la leucemia
Para el público general, el mensaje es que las células leucémicas pueden escapar a un fármaco dirigido no solo al mutar su diana directa, sino también al cambiar cómo producen y usan energía. El estudio muestra que las células de LMA resistentes a cabozantinib adoptan una estrategia de “quemar azúcar” mientras dejan decaer sus mitocondrias. Al atacar las vías que sostienen este metabolismo reprogramado —a través de fármacos como omipalisib o inhibidores de HSP90— podría ser posible restaurar la sensibilidad al cabozantinib y tratamientos similares. Aunque estos hallazgos provienen de modelos celulares y no de pacientes, sugieren que combinar fármacos dirigidos con agentes que modulen el metabolismo podría ser una vía prometedora para retrasar o superar la resistencia en LMA con mutación FLT3.
Cita: Fu, YH., Ng, K.M., Tseng, CY. et al. Modulating metabolic signatures to mitigate cabozantinib resistance in FLT3-ITD acute myeloid leukemia cell models. Cell Death Discov. 12, 98 (2026). https://doi.org/10.1038/s41420-026-02957-8
Palabras clave: leucemia mieloide aguda, resistencia a fármacos, mutación FLT3, metabolismo del cáncer, cabozantinib