Clear Sky Science · es
La inhibición de RRM1 sensibiliza el adenocarcinoma de pulmón al tratamiento con decitabina
Convertir un fármaco tibio en un aliado más potente
El cáncer de pulmón sigue siendo uno de los más letales y muchos pacientes acaban quedándose sin opciones terapéuticas efectivas. Los médicos llevan tiempo esperando que fármacos que reprograman suavemente el ADN de las células cancerosas, en lugar de limitarse a envenenar las células en división, puedan ayudar. Uno de esos fármacos, la decitabina, funciona bien en cánceres de la sangre pero ha decepcionado en tumores sólidos como el de pulmón. Este estudio plantea una pregunta simple y práctica con grandes implicaciones: ¿existe una forma de lograr que los tumores pulmonares respondan a la decitabina, utilizando herramientas que ya conocemos?
Por qué un fármaco probado falla en tumores sólidos
La decitabina es un análogo de uno de los bloques constructores del ADN. Cuando se incorpora al ADN de una célula durante la replicación, puede borrar marcas químicas anormales que silencian genes protectores, incluidos supresores de tumores y genes inmunitarios. En las leucemias, esto ayuda a reorientar las células hacia un estado más saludable. Sin embargo, en los tumores de pulmón el fármaco apenas funciona. Los autores sospecharon que el problema no era lo que hace la decitabina, sino la escasa cantidad que realmente llega al ADN de las células de tumores sólidos. Midiendo minúsculas cantidades de fármaco incorporadas en el ADN en varias líneas celulares cancerosas, confirmaron que las células que incorporaban más decitabina eran mucho más sensibles a su efecto letal.
Un portero celular que bloquea el fármaco
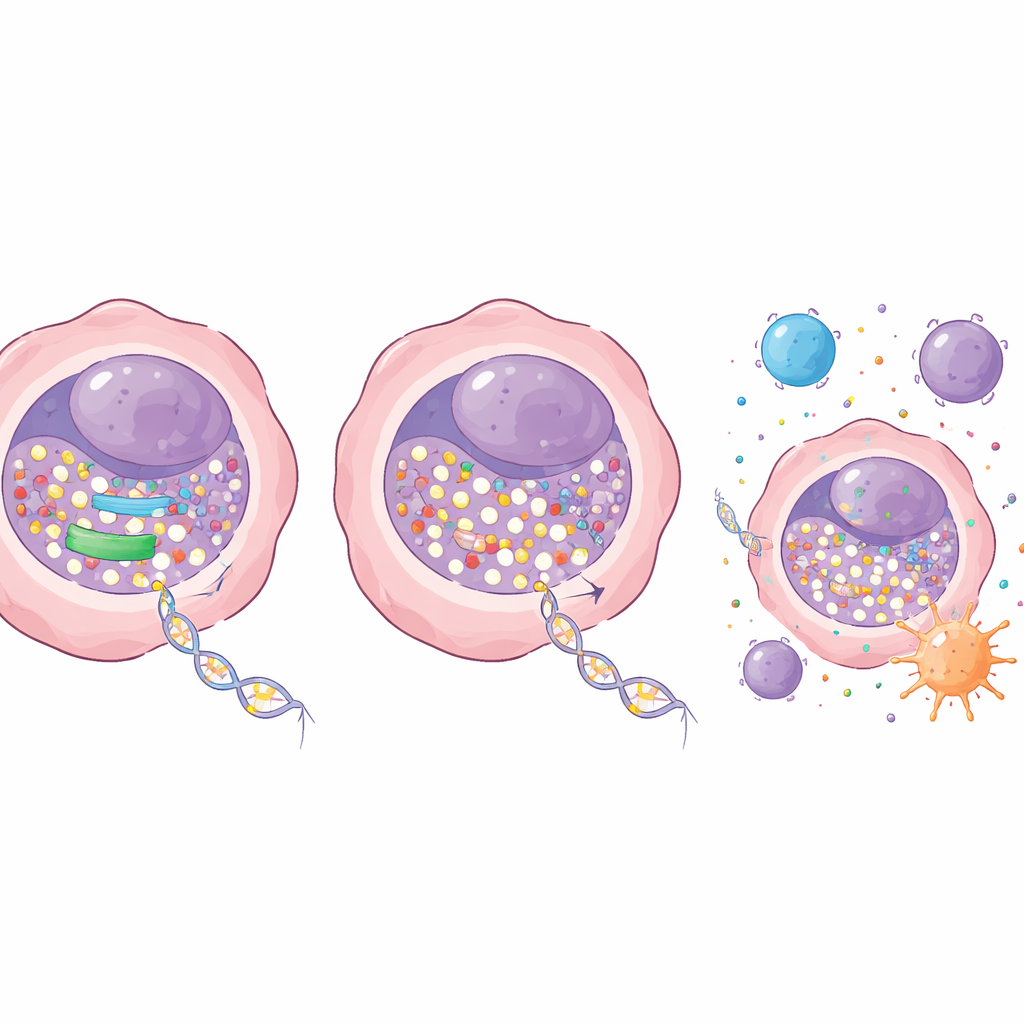
Para averiguar qué limita la entrada del fármaco en el ADN, los investigadores examinaron genes implicados en el manejo de nucleósidos —las materias primas del ADN. Una enzima, llamada RRM1, destacó. RRM1 forma parte de una maquinaria que convierte los bloques ordinarios en las formas activas usadas para fabricar ADN. En el adenocarcinoma de pulmón, esta enzima era inusualmente abundante en los tumores comparada con el tejido pulmonar normal, y los pacientes con niveles más bajos de RRM1 tendían a vivir más. En un panel de líneas celulares cancerosas, niveles más altos de RRM1 se asociaron con una menor incorporación de decitabina, lo que sugiere con fuerza que esta enzima actúa como un portero que desplaza al fármaco.
Desarmar al portero para ayudar al fármaco
El equipo preguntó luego qué sucede si se desactiva parcialmente RRM1. Usando herramientas genéticas, redujeron la expresión de RRM1 en células de cáncer de pulmón sin matar las células de forma directa. Por sí sola, esta reducción tuvo solo un efecto leve en el crecimiento. Pero combinada con dosis bajas de decitabina, el impacto fue drástico: las colonias de células cancerosas de pulmón se redujeron notablemente en placas de cultivo y los tumores crecieron mucho más despacio en ratones. Importa que estas dosis efectivas fueron bien toleradas, sin daños evidentes en sangre, hígado o riñón en los animales. A nivel molecular, bloquear RRM1 permitió que se incorporara más decitabina al ADN, provocando una pérdida más marcada de la enzima que añade metilos, DNMT1, y una mayor caída de la metilación global del ADN. Esto, a su vez, reactivó genes supresores de tumores que estaban silenciados.
Activar la alarma inmune dentro de los tumores
Más allá de ralentizar la división celular, la terapia combinada modificó la interacción de las células cancerosas con el sistema inmunitario. La mayor presencia de decitabina en el ADN aumentó las señales de daño en el ADN dentro de las células y las impulsó hacia la muerte celular programada. Al mismo tiempo, potenció la actividad de un sistema de alarma interno centrado en la vía STING, que detecta ADN fuera de lugar y desencadena respuestas inmunes similares a las antivirales. Cuando se bloqueó RRM1, la decitabina activó con más fuerza esta vía y sus genes diana, incluidos aquellos que reclutan y estimulan células inmunitarias. En modelos murinos de cáncer de pulmón con sistemas inmunitarios intactos, combinar decitabina con un inhibidor de RRM1 produjo un control tumoral superior al de cualquiera de los tratamientos por separado, sin añadir toxicidad evidente. Los autores también encontraron que esta estrategia de inhibición potencia específicamente la decitabina y puede, de hecho, actuar en sentido contrario con un fármaco relacionado, la azacitidina, lo que subraya la necesidad de emparejamientos adecuados.
Qué podría significar esto para los pacientes
En conjunto, el trabajo dibuja una imagen clara: una enzima de síntesis de ADN hiperactiva en los tumores pulmonares limita la cantidad de decitabina que alcanza su objetivo. Al bloquear parcialmente esta enzima, las células cancerosas se ven obligadas a utilizar más del fármaco en lugar de sus bloques habituales. Ese cambio permite que dosis bajas de decitabina reactiven con más eficacia genes protectores, dañen el ADN de las células tumorales y despierten defensas inmunitarias, todo ello manteniendo tolerabilidad en modelos animales. Para los pacientes, esto sugiere una vía realista: reutilizar o perfeccionar inhibidores de la enzima RRM1, en combinación con decitabina a baja dosis y, potencialmente, con inmunoterapias modernas, para convertir un fármaco hasta ahora poco eficaz en un componente útil del tratamiento del cáncer de pulmón.
Cita: Jiang, N., Liu, J., Vaghasia, A. et al. RRM1 inhibition sensitizes lung adenocarcinoma to decitabine treatment. Cell Death Dis 17, 275 (2026). https://doi.org/10.1038/s41419-026-08522-6
Palabras clave: adenocarcinoma de pulmón, decitabina, metilación del ADN, ribonucléotido reductasa, inmunoterapia contra el cáncer