Clear Sky Science · es
Las variantes de pérdida de función en HPDL alteran el desarrollo cortical humano mediante cambios en la función mitocondrial
Por qué importan los pequeños motores celulares para cerebros en crecimiento
La mayoría de la gente piensa en la genética y el cableado al imaginar cómo se desarrolla el cerebro. Este estudio muestra que otro factor, con frecuencia pasado por alto —las pequeñas centrales eléctricas dentro de nuestras células llamadas mitocondrias— también puede moldear la formación cerebral. Al estudiar trastornos infantiles raros del movimiento vinculados a un gen llamado HPDL, los autores revelan cómo una producción de energía defectuosa puede reducir la corteza en desarrollo, la región cerebral crucial para el movimiento, el pensamiento y el comportamiento.
Un raro trastorno del movimiento como ventana al crecimiento cerebral
Algunos niños con alteraciones en el gen HPDL desarrollan paraparesia espástica hereditaria, una condición que provoca rigidez y debilidad en las piernas, junto con convulsiones, retraso del desarrollo y, en los casos más graves, un cerebro de menor tamaño de lo normal (microcefalia). Aunque se sabía que la proteína HPDL se localiza en las mitocondrias, su función exacta —y por qué su pérdida daña el cerebro— no estaba clara. Los investigadores usaron varios modelos celulares humanos, incluidas células tumorales con apariencia nerviosa y células cerebrales obtenidas a partir de muestras de piel de pacientes, para probar si HPDL es necesario para el desarrollo cerebral normal y la salud mitocondrial.
Qué ocurre cuando HPDL se apaga
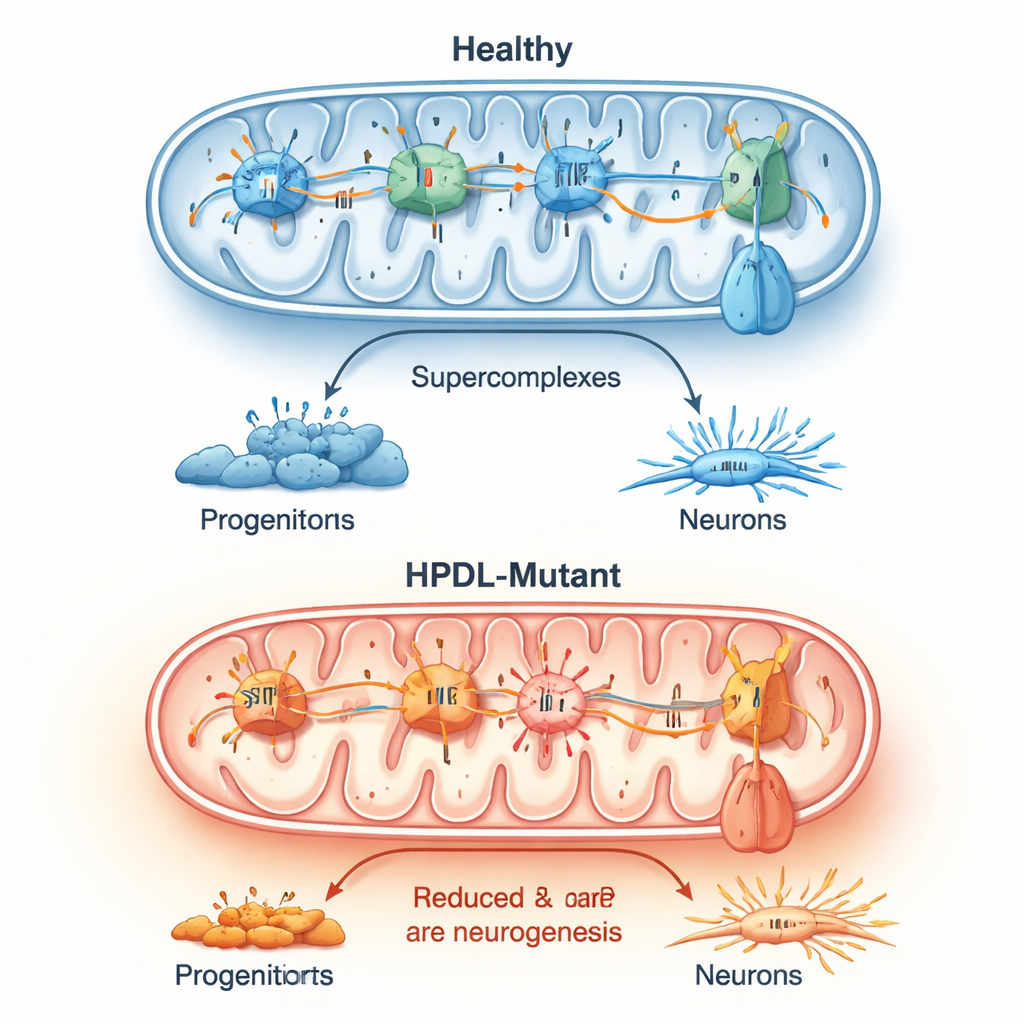
Primero, el equipo inactivó HPDL en una línea celular humana de neuroblastoma mediante edición génica CRISPR. Sin HPDL, estas células perdieron la proteína de longitud completa y mostraron problemas mitocondriales claros. Los grandes ensamblajes de proteínas de la cadena respiratoria que normalmente trabajan juntos para generar energía se vieron alterados, y componentes clave implicados en el uso del oxígeno se redujeron. Las células consumieron menos oxígeno, produjeron menos respiración ligada a la energía y generaron más especies reactivas de oxígeno —subproductos dañinos a menudo denominados “estrés oxidativo”. Sin embargo, el número total de mitocondrias no disminuyó, y los niveles de coenzima Q10, una molécula vital para la transferencia de energía, fueron en realidad más altos, lo que sugiere un defecto cualitativo —no solo cuantitativo— en la función mitocondrial.
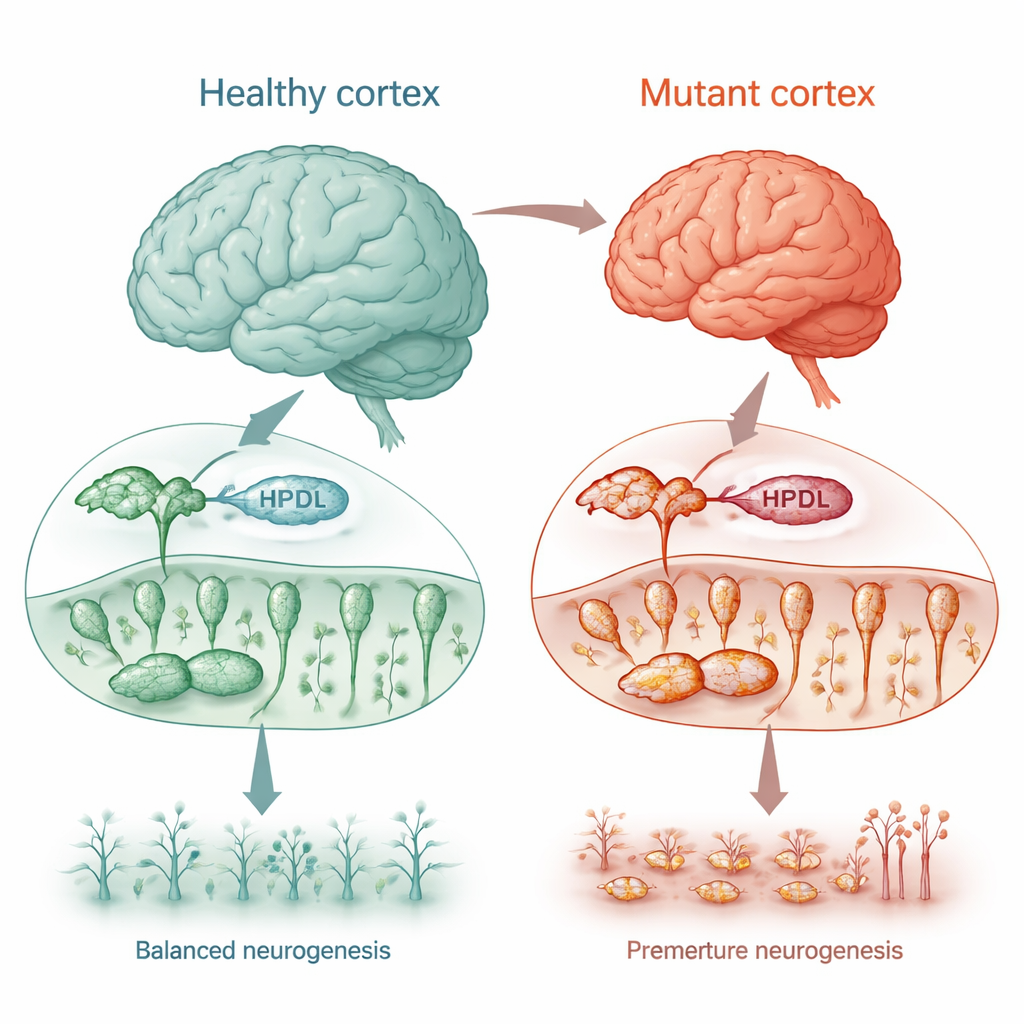
Tejido cerebral en placa revela una sobreproducción temprana de neuronas
Para ver cómo la pérdida de HPDL afecta el verdadero desarrollo cerebral humano, los investigadores reprogramaron células de piel de cuatro niños afectados en células madre pluripotentes inducidas y luego las indujeron a formar células corticales y “mini-cerebros” tridimensionales (organoides). Temprano en el desarrollo, en una etapa en la que la mayoría de las células aún deberían dividirse como progenitores neuronales, las culturas con mutación en HPDL ya contenían más neuronas maduras y menos progenitores. Los perfiles de actividad génica confirmaron esto: las vías que impulsan la formación neuronal se activaron prematuramente, mientras que las que mantienen a las células en un estado proliferativo se atenuaron. En los organoides, este cambio prematuro de bloques constructores a neuronas terminadas condujo a estructuras cerebrales mucho más pequeñas, reflejando la microcefalia observada en los niños más graves.
Plantas de energía rotas y células estresadas
Una inspección más detallada mostró que las células cerebrales con mutación en HPDL tenían comprometida la fosforilación oxidativa —la vía principal por la que las mitocondrias generan energía. Tinciones enzimáticas revelaron una actividad más débil de un complejo mitocondrial clave, mientras que otras mediciones mostraron alteraciones en la diferencia de potencial a través de la membrana mitocondrial. En muchas células mutantes, una enzima crucial que normalmente sintetiza ATP parecía funcionar en sentido inverso para sostener este potencial de membrana, una señal de grave estrés metabólico. En las líneas de pacientes, las especies reactivas de oxígeno estaban consistentemente elevadas y los grandes ensamblajes de proteínas de la cadena respiratoria estaban menos bien formados. Estos cambios mitocondriales se correlacionaron estrechamente con el momento y el grado de la producción neuronal prematura.
Probar maneras de aliviar el estrés
Dado que el estrés oxidativo y la alteración de la química de la coenzima Q10 parecían centrales, el equipo probó si tratamientos dirigidos a estos problemas podían frenar la aceleración hacia la formación neuronal. Expusieron culturas corticales tempranas a dos antioxidantes y a 4‑hidroxibenzato, una pequeña molécula relacionada con la síntesis de coenzima Q10. En varias líneas derivadas de pacientes, estos compuestos redujeron en parte la neurogénesis prematura, pero la respuesta dependió de la mutación exacta de HPDL. Algunas líneas respondieron principalmente a los antioxidantes, otras al precursor de la coenzima Q10, y una no respondió en absoluto. Este patrón específico de mutación sugiere que podrían necesitarse estrategias de tratamiento personalizadas para los trastornos relacionados con HPDL.
Qué significa esto para los niños y las futuras terapias
En términos simples, este estudio muestra que HPDL actúa como guardián de los bloques constructores del cerebro durante el desarrollo temprano. Cuando HPDL falla, las mitocondrias se vuelven ineficientes y excesivamente estresadas, empujando a las células progenitoras a convertirse en neuronas demasiado pronto. La reserva de células en división se agota, la corteza no alcanza su tamaño completo y los patrones de conectividad se alteran, contribuyendo a problemas de movimiento y otros síntomas. La mejora parcial observada con antioxidantes y compuestos relacionados con la coenzima Q10 sugiere que ajustar el equilibrio energético celular y el estrés oxidativo podría algún día ayudar a niños con mutaciones en HPDL, y quizás a otros con formas mitocondriales de enfermedad cerebral.
Cita: Baggiani, M., Desbats, M.A., Naef, V. et al. Loss of function variants in HPDL impair human cortical development via alterations of mitochondrial function. Cell Death Dis 17, 237 (2026). https://doi.org/10.1038/s41419-026-08476-9
Palabras clave: HPDL, mitocondrias, desarrollo cortical, microcefalia, estrés oxidativo