Clear Sky Science · es
Mantenimiento aberrante del factor de transcripción del desarrollo PAX6 promueve la muerte neuronal vía señalización JNK3
Por qué esta investigación importa para la visión
El glaucoma es una causa principal de ceguera irreversible, en gran parte porque las células nerviosas que transportan la información visual desde el ojo hasta el cerebro mueren de forma gradual. Muchos tratamientos reducen la presión intraocular, pero las personas aún pueden perder la vista incluso cuando la presión está bien controlada. Este estudio plantea una pregunta más profunda: ¿qué hace que estas células nerviosas retinianas decidan morir cuando están bajo estrés, y podemos apagar esa decisión a nivel del control genético dentro del núcleo celular?
Una retina estresada bajo ataque
En el corazón del glaucoma y enfermedades oculares relacionadas está la pérdida lenta de las células ganglionares retinianas (CGR), las neuronas de salida del ojo. Estas células son vulnerables a muchos tipos de estrés, incluido el exceso tóxico del neurotransmisor glutamato, que sobreactiva los receptores NMDA y desencadena una dañina sobrecarga de calcio. Los investigadores utilizaron un modelo bien establecido en ratón en el que se inyecta una pequeña cantidad de NMDA en el ojo, lesionando selectivamente las CGR mientras deja otras capas retinianas mayormente intactas. Confirmaron que este tratamiento no alteró la presión ocular, pero sí provocó signos típicos de muerte celular programada en las CGR, como la liberación de citocromo c desde las mitocondrias y la aparición de núcleos TUNEL-positivos.
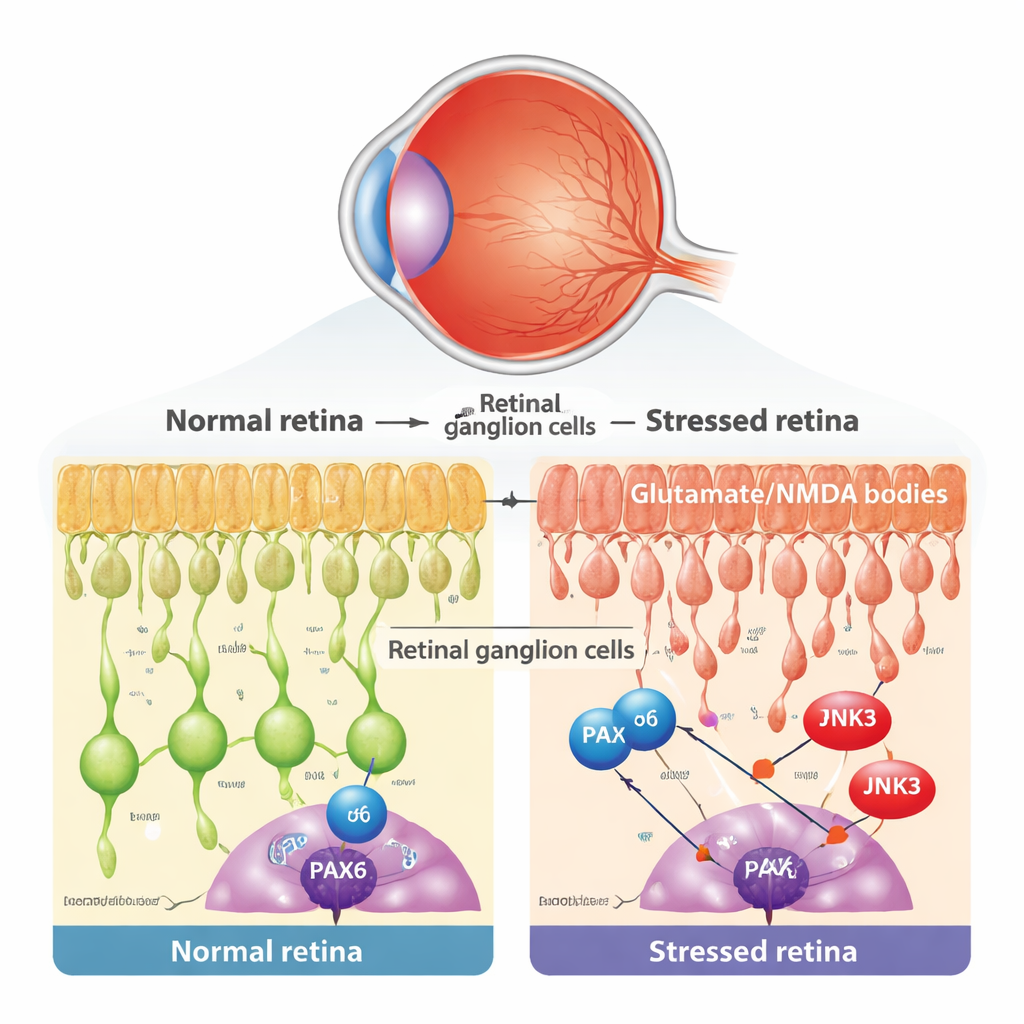
Un gen del desarrollo que se niega a retirarse
Durante el desarrollo temprano, un regulador génico llamado PAX6 actúa como arquitecto maestro del ojo, guiando cómo nacen y se conectan las distintas células retinianas. La sabiduría convencional sostiene que esos programas de desarrollo se apagan en la edad adulta. Al reanalizar datos de secuenciación de ARN unicelular de retinas de ratón y humanas, el equipo encontró que PAX6, de hecho, se mantiene de manera fuerte y selectiva en CGR maduras y en ciertas interneuronas. Usando tinción microscópica, mostraron que en la capa donde residen las CGR, PAX6 está mayormente presente en las células ganglionares más que en las células amacrinas vecinas. Esto planteó una posibilidad intrigante: en la enfermedad adulta, un antiguo programa de desarrollo podría ser secuestrado y convertirse en un impulsor de la degeneración.
De guardián a verdugo: PAX6 cambia de papel
Para probar si PAX6 ayuda a las CGR a sobrevivir o a morir bajo estrés, los científicos emplearon un enfoque parecido a la terapia génica. Administraron un vector viral que lleva un ARN pequeño que reduce específicamente PAX6 en la retina, y luego expusieron los ojos a NMDA. En comparación con los ojos tratados con control, las retinas con PAX6 disminuido mostraron muchas menos CGR apoptóticas y mucho menos daño mitocondrial, lo que indica que PAX6 es necesario para la muerte celular completa en este modelo. La secuenciación de ARN a nivel genómico reveló que muchos genes pro-muerte, particularmente los implicados en daño mitocondrial y activación de caspasas, se indujeron fuertemente por NMDA en ratones normales pero se vieron atenuados cuando PAX6 fue silenciado. En otras palabras, PAX6 ayuda a activar una red de genes que empuja a las CGR al abismo.
La quinasa de estrés que activa el interruptor PAX6
¿Cómo activa el estrés a PAX6 sin aumentar su cantidad? El equipo se centró en JNK3, una enzima sensible al estrés que se encuentra principalmente en neuronas. Bajo lesión por NMDA, JNK3 se trasladó al núcleo de las CGR y se asoció físicamente con PAX6. Experimentos bioquímicos "de tubo de ensayo" con proteínas purificadas mostraron que JNK3 puede añadir directamente grupos fosfato a PAX6, y esta reacción fue bloqueada por un inhibidor de JNK. En ratones carentes del gen Jnk3, NMDA ya no produjo el mismo patrón de fosforilación de PAX6. El mapeo de la cromatina (ChIP-seq) y ensayos dirigidos de unión al ADN revelaron que, bajo estrés, PAX6 fosforilado, junto con JNK3, se une con mayor fuerza a las regiones de control de genes proapoptóticos clave como Bax y Gadd45a, potenciando su actividad. Cuando PAX6 fue reducido o JNK3 fue eliminado genéticamente, esta unión y la correspondiente activación de genes pro-muerte se redujeron drásticamente.
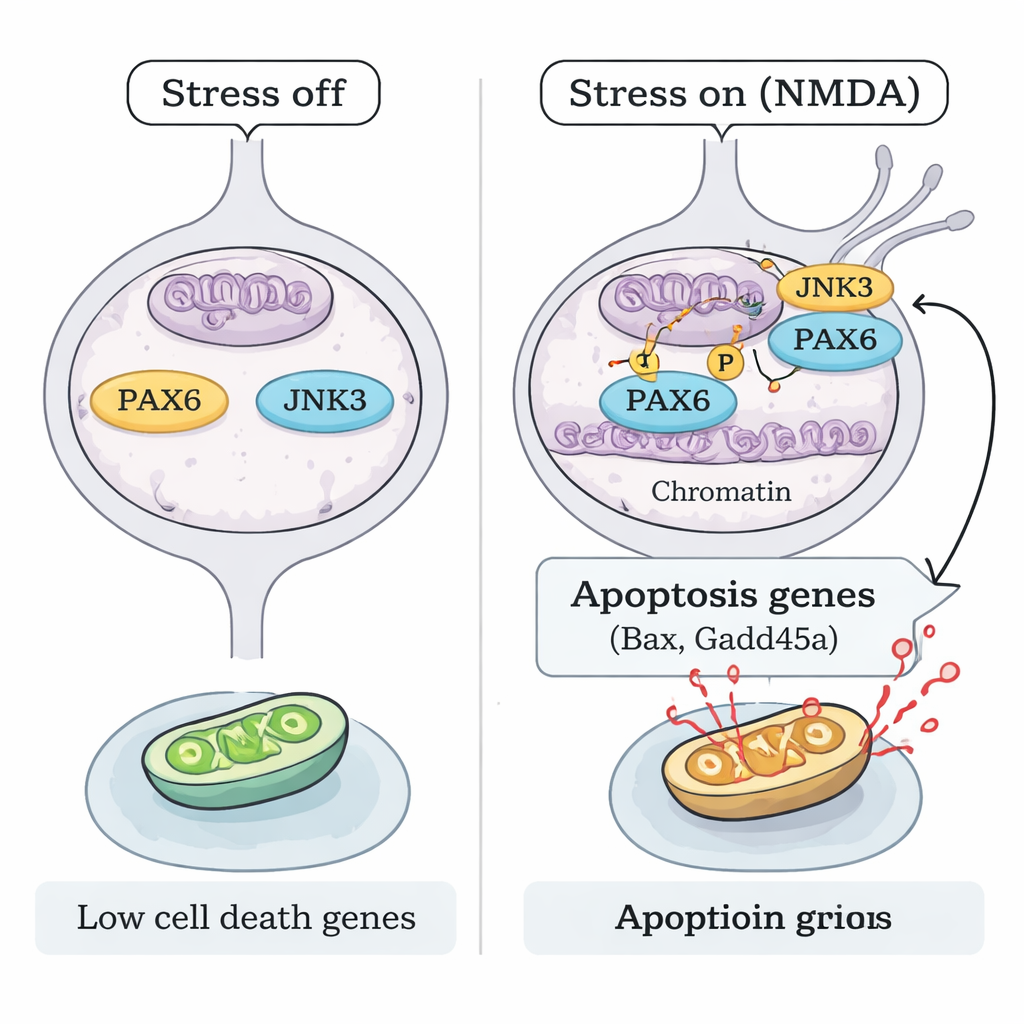
Apagar el programa de muerte para proteger la visión
Finalmente, los investigadores preguntaron si bloquear este eje JNK3–PAX6 basta para proteger las células críticas para la visión. Tanto en ratones con reducción de PAX6 como en ratones deficientes en JNK3, las CGR se conservaron de forma significativa tras la exposición a NMDA, con menos células muertas y una estructura retiniana más saludable. Esto apunta a un modelo mecanístico claro: bajo estrés excitotóxico, JNK3 fosforila el PAX6 persistentemente expresado, convirtiéndolo de un constructor del desarrollo en un potente activador de un programa génico de muerte celular en las CGR adultas. Interrumpir ese vínculo —silenciando PAX6 o deshabilitando JNK3— mantiene vivas a muchas de estas neuronas. Para los pacientes, este trabajo sugiere que futuras terapias para el glaucoma podrían ir más allá de bajar la presión ocular y dirigirse directamente a los interruptores genéticos que deciden si las neuronas retinianas viven o mueren.
Cita: Kim, JY., An, MJ., Kim, J. et al. Aberrant maintenance of developmental transcription factor PAX6 promotes neuronal cell death via JNK3 signaling. Cell Death Dis 17, 161 (2026). https://doi.org/10.1038/s41419-026-08417-6
Palabras clave: glaucoma, células ganglionares retinianas, PAX6, JNK3, neurodegeneración