Clear Sky Science · es
La activación de la señalización del factor de crecimiento nervioso limita la respuesta al lenvatinib en el carcinoma hepatocelular
Por qué importa esta historia sobre el cáncer
El cáncer de hígado es uno de los más letales a nivel mundial, y aun con fármacos modernos muchos pacientes dejan de responder después de un periodo inicial de beneficio. Este estudio indaga por qué un medicamento muy usado, el lenvatinib, a menudo pierde eficacia frente a tumores hepáticos avanzados. Los investigadores descubren un cómplice inesperado procedente de la biología nerviosa—el factor de crecimiento nervioso—y muestran cómo bloquear esta señal podría ayudar a que los tratamientos actuales funcionen durante más tiempo y con mayor eficacia.
Cuando un fármaco útil se queda sin efecto
El lenvatinib es una pastilla que frena el crecimiento tumoral cortando señales de crecimiento y el suministro de sangre. Se ha convertido en un pilar para personas con cáncer de hígado inoperable. Sin embargo, la mayoría de los tumores finalmente “aprenden” a convivir con el fármaco, y la supervivencia de los pacientes no ha mejorado tanto como se esperaba. Para estudiar este problema en condiciones realistas, el equipo hizo crecer tumores humanos de hígado en ratones, los trató con lenvatinib y luego trasladó repetidamente las células tumorales supervivientes entre animales y cultivos. Tras varios ciclos, crearon poblaciones celulares extremadamente difíciles de eliminar con el fármaco, recreando de cerca la resistencia observada en la clínica.
Una señal nerviosa que alimenta el tumor en secreto
Usando el líquido que rodeaba estas células resistentes como pista, los investigadores buscaron proteínas que las células liberaban a su entorno. Una molécula destacó: el factor de crecimiento nervioso (NGF), conocido por guiar el crecimiento y la supervivencia de las neuronas. A medida que las células se volvían más resistentes, secretaban progresivamente más NGF. Cuando este líquido rico en NGF se añadió a células previamente sensibles, esas células también se volvieron más difíciles de matar con lenvatinib. Añadir NGF purificado por sí solo fue suficiente para atenuar el efecto del fármaco, mientras que otros factores de crecimiento no tuvieron el mismo impacto. Eliminar NGF en células resistentes restauró su vulnerabilidad al tratamiento y ralentizó el crecimiento tumoral en ratones, especialmente bajo tratamiento con lenvatinib. En muestras de pacientes, los tumores que persistieron o reaparecieron tras la terapia con lenvatinib mostraron niveles mucho más altos de NGF que los tumores no tratados, y los pacientes con tumores con NGF elevado tuvieron peor supervivencia.
Cómo las células tumorales reconfiguran su maquinaria interna
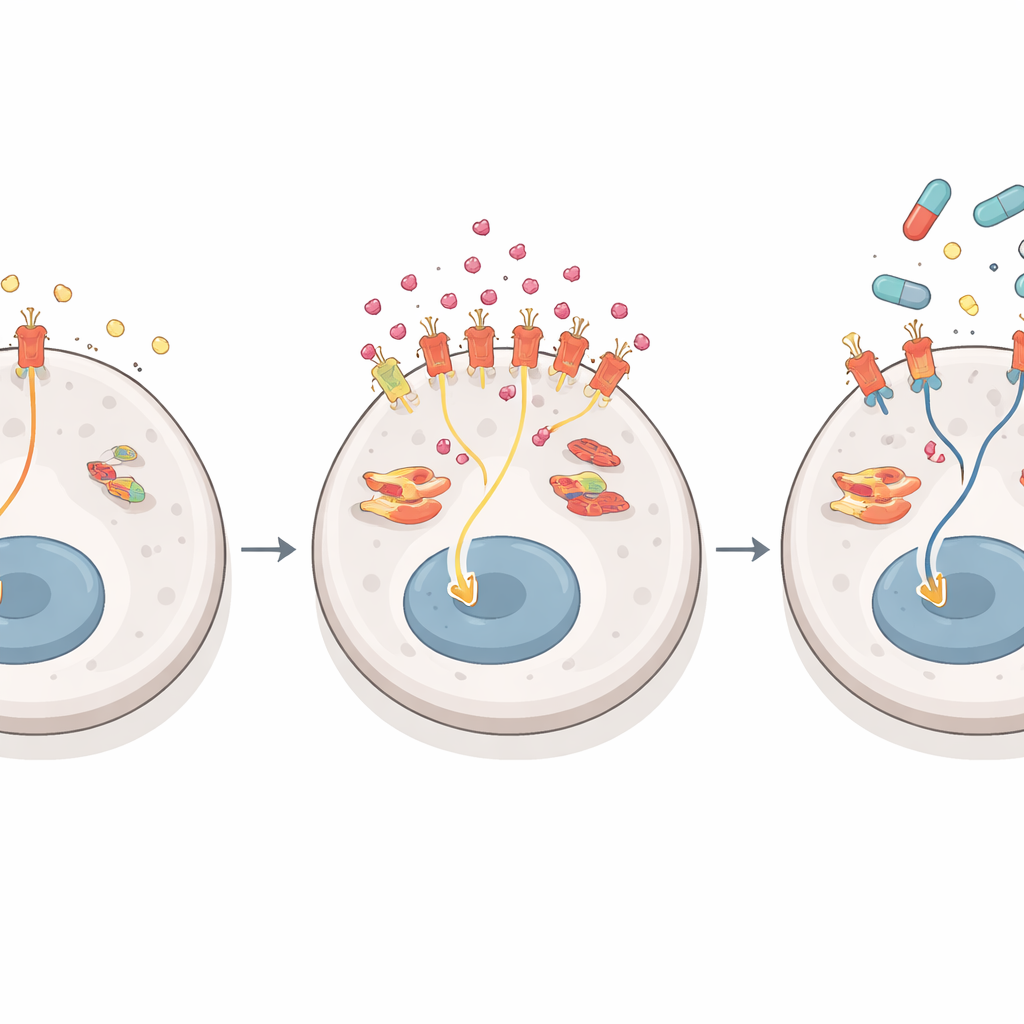
El equipo preguntó a continuación cómo las células de cáncer hepático aumentan la producción de NGF sin cambiar el gen subyacente ni ralentizar su degradación. Encontraron la respuesta en cómo las células cortan y ensamblan el plano de ARN del NGF. El gen NGF puede procesarse en una versión mensajera larga o corta. En células sensibles al fármaco predomina la forma larga; en las resistentes, la forma corta se impone y se traduce a proteína con mucha mayor eficiencia. Una proteína de empalme llamada SRSF1 se une específicamente a la región del ARN que define esta forma corta. Su actividad, a su vez, es potenciada por una quinasa llamada SRPK1, que añade grupos fosfato y ayuda a transportar SRSF1 al núcleo celular, donde ocurre el empalme. En las células resistentes, SRPK1 está elevado, SRSF1 se vuelve más activo en el núcleo y el equilibrio se inclina hacia la versión de ARN de alto rendimiento de NGF, impulsando una oleada en la liberación de proteína NGF.
Un conmutador de señal que elude el fármaco
El NGF actúa uniéndose a un receptor en las células tumorales llamado TrkA. Cuando TrkA se activa en células resistentes, redirige el flujo de señales de crecimiento dentro de la célula. En condiciones normales, las células de cáncer hepático dependen principalmente de una cadena clásica de proteínas—a menudo llamada vía ERK1/2—para impulsar el crecimiento. El lenvatinib es muy eficaz en interrumpir esta vía principal. Pero en células resistentes literalmente inundadas de NGF, TrkA favorece una cadena paralela que termina en una proteína llamada ERK5. A medida que el lenvatinib cierra la vía habitual, el tumor cambia silenciosamente su dependencia hacia la vía ERK5, manteniendo activas las señales de crecimiento y supervivencia. Bloquear TrkA o ERK5 en combinación con lenvatinib hizo que las células resistentes fuesen mucho más sencillas de eliminar en ensayos de crecimiento a largo plazo, mientras que tuvo poco efecto adicional en células sensibles al fármaco. En las etapas iniciales de resistencia, otras señales como las del receptor de EGF parecen más importantes, pero a medida que la resistencia se afianza, la ruta NGF–TrkA–ERK5 se convierte en la vía de escape dominante.
Convertir una debilidad en un nuevo plan de tratamiento
Puesto que SRPK1 tiene muchos roles en células sanas, los autores se centraron en TrkA como un objetivo más práctico. Probaron larotrectinib, un fármaco ya aprobado para ciertos tumores impulsados por fusiones del gen TRK. En modelos de ratón diseñados para sobreproducir SRPK1 en el hígado, el lenvatinib por sí solo apenas ralentizó los tumores una vez que aumentaron los niveles de NGF, mientras que el larotrectinib solo tuvo un beneficio modesto. La combinación, sin embargo, redujo fuertemente los tumores sin toxicidad añadida evidente. En injertos tumorales derivados de pacientes y mini-tumores cultivados a partir de individuos cuyos cánceres se habían vuelto resistentes al lenvatinib con niveles altos de NGF, el larotrectinib restauró la sensibilidad al lenvatinib y la pareja funcionó mucho mejor que cualquiera de los fármacos por separado. En contraste, los tumores con NGF bajo siguieron controlándose bien con lenvatinib solo y obtuvieron poco beneficio adicional al añadir larotrectinib.
Qué significa esto para los pacientes
Este trabajo muestra que algunos cánceres de hígado escapan al lenvatinib activando un bucle de crecimiento de tipo nervioso: SRPK1 y SRSF1 remodelan el ARN de NGF, aumentando la producción de NGF; el NGF activa entonces TrkA y reconfigura el cableado interno del tumor hacia una vía de respaldo que el lenvatinib no bloquea bien. De forma alentadora, esta misma reconfiguración revela una nueva vulnerabilidad. Usar un fármaco ya existente que bloquee TrkA junto con lenvatinib—especialmente en pacientes cuyos tumores muestran NGF alto o TrkA activo—podría volver a sensibilizar a los cánceres resistentes manteniéndose dentro de límites de seguridad ya conocidos. Si se confirma en ensayos clínicos, una prueba sencilla de tejido para NGF o actividad de TrkA podría guiar a los médicos hacia un enfoque combinado y más personalizado para personas con cáncer de hígado avanzado.
Cita: Xu, M., Zheng, Y., Zhao, L. et al. Activation of Nerve Growth Factor signaling limits the response to lenvatinib in hepatocellular carcinoma. Sig Transduct Target Ther 11, 120 (2026). https://doi.org/10.1038/s41392-026-02649-w
Palabras clave: carcinoma hepatocelular, resistencia a fármacos, factor de crecimiento nervioso, terapia dirigida, lenvatinib