Clear Sky Science · es
Potenciar la actividad de KLF15 en cardiomiocitos: un enfoque novedoso para prevenir la reprogramación patológica y la fibrosis mediante dCas9VPR sin nuclease
Reprogramando el corazón en fallo
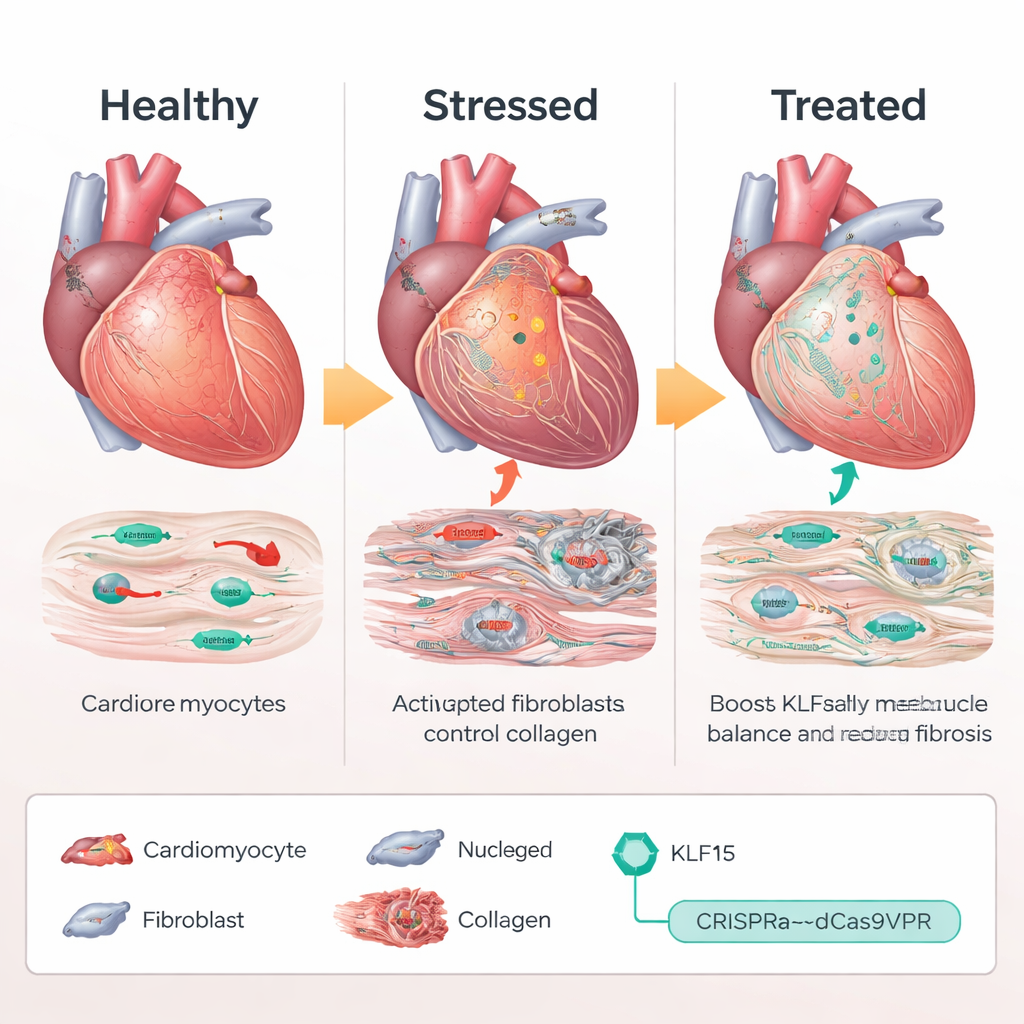
La insuficiencia cardiaca afecta a millones de personas y a menudo se desarrolla de forma gradual tras años de hipertensión o enfermedad valvular. En estas condiciones, las células del músculo cardiaco no solo aumentan de tamaño, sino que activan un programa genético «fetal» y el corazón se llena de tejido cicatricial. Este estudio explora una nueva manera de llevar la maquinaria de control génico del propio corazón de vuelta hacia la salud—sin cortar el ADN—al aumentar con delicadeza un regulador protector llamado KLF15 en las células del músculo cardiaco.
Cuando las células cardíacas pierden su identidad
En un corazón adulto sano, los cardiomiocitos—las células del músculo cardiaco—queman lípidos de forma eficiente para obtener energía y mantienen un patrón estable de actividad génica. Mediante secuenciación de ARN unicelular en ratones sometidos a sobrecarga de presión crónica, los investigadores cartografiaron cómo cambian las cardiomiocitos individuales a medida que el corazón pasa de función normal a dilatación y, finalmente, a fallo. Encontraron que un factor de transcripción llamado KLF15, que normalmente mantiene el equilibrio entre metabolismo y crecimiento, mostraba el cambio de actividad más pronunciado en las células enfermas. A medida que aumentaba el estrés, los niveles de KLF15 caían y su capacidad para controlar los genes fetales y relacionados con el estrés se debilitaba. Descensos similares de KLF15 se observaron en corazones humanos de pacientes con miocardiopatía dilatada e hipertrófica, lo que indica que esta alteración se conserva entre especies.
Usar CRISPR como un mando de volumen, no como unas tijeras
En lugar de añadir una copia extra del gen KLF15 o cortar el ADN, el equipo empleó un sistema CRISPR de «activación», llamado dCas9VPR, que se une cerca del gen Klf15 natural y potencia su propia expresión. En ratones diseñados para expresar este activador CRISPR solo en cardiomiocitos, los científicos entregaron ARN guía mediante un virus adenoasociado (AAV9) para dirigir el promotor de Klf15. Bajo sobrecarga de presión crónica, los ratones que recibieron guías activadoras de Klf15 mantuvieron niveles cercanos a lo normal de Klf15. Sus cardiomiocitos permanecieron más pequeños, la función de bombeo se deterioró menos y la supervivencia mejoró en comparación con animales control. A nivel molecular, los genes de estrés y fetales se silenciaron, mientras que los genes metabólicos y los relacionados con el manejo del calcio se recuperaron, lo que indica que el programa transcripcional patológico se había restablecido en gran medida.
Silenciar la formación de cicatriz mediante la comunicación entre células
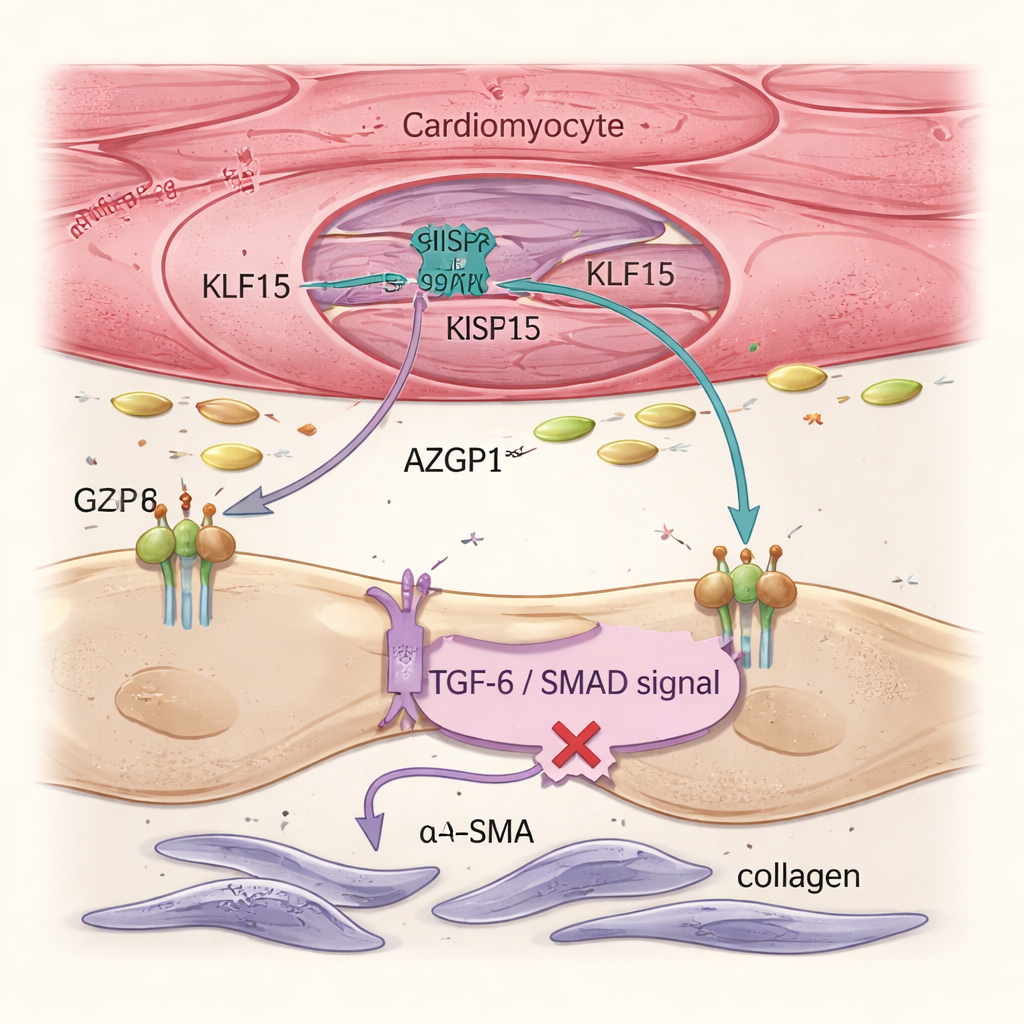
La insuficiencia cardiaca no la impulsan solo las células musculares enfermas, sino también los fibroblastos, células de soporte que producen colágeno y forman tejido cicatricial rígido. Los análisis unicelulares y la imagen tisular mostraron que restaurar Klf15 en cardiomiocitos redujo la activación de fibroblastos y la fibrosis global, aunque la terapia génica nunca se dirigió directamente a los fibroblastos. El equipo rastreó este efecto hasta una proteína secretada llamada AZGP1. Cuando Klf15 se incrementó en cardiomiocitos, la producción y liberación de AZGP1 aumentó. Tanto en corazones de ratón como en tejidos cardíacos humanos derivados de células madre, niveles más altos de AZGP1 atenuaron la vía TGF-β / SMAD en fibroblastos—un motor clave de la cicatrización—reduciendo marcadores como α-SMA y POSTN. Es importante señalar que la sobreexpresión de AZGP1 solo en cardiomiocitos no reprogramó las células musculares, lo que demuestra que KLF15 protege principalmente a los cardiomiocitos de forma directa y emplea AZGP1 como mensajero para frenar a los fibroblastos.
Modelos de tejido humano confirman el circuito protector
Para comprobar si estos mecanismos se mantienen en células humanas, los investigadores usaron cardiomiocitos derivados de células madre pluripotentes inducidas, cultivados en tejidos cardíacos tridimensionales de ingeniería. Cuando se sometieron a carga mecánica que imita la hipertensión, estos tejidos perdieron KLF15, activaron genes de estrés y fetales, se rigidizaron y sus contracciones se debilitaron—recapitulando rasgos de la enfermedad. La restauración de KLF15 mediante CRISPRa previno este declive, preservó la generación de fuerza y desplazó la expresión génica hacia un metabolismo y una estructura más madura. Experimentos detallados mostraron que TGF-β1, una señal profibrótica bien conocida, reduce KLF15 en cardiomiocitos humanos a través de su vía SMAD2/3, lo que ayuda a explicar cómo el estrés crónico conduce a una remodelación maladaptativa. Finalmente, el equipo diseñó un sistema CRISPRa compacto, «mini», basado en una variante de Cas9 más pequeña que cabe en un único vector AAV9 y está impulsado por un promotor específico de cardiomiocitos. En cortes precisos de tejido cardiaco humano en fallo, este vector elevó con éxito los niveles de KLF15 y mejoró el rendimiento contráctil durante días en cultivo.
Un plan para una terapia génica más suave
Para un lector no especializado, el mensaje central es que este trabajo muestra cómo aumentar con cuidado un único regulador protector dentro de las células del músculo cardiaco puede tanto estabilizar su identidad como enviar señales que limitan la cicatrización. Al usar un activador basado en CRISPR que no corta el ADN, el enfoque ajusta el gen propio del corazón en lugar de insertar uno artificial. El estudio define una vía TGF-β → KLF15 → AZGP1 que enlaza el estrés mecánico con la remodelación dañina y demuestra, en ratones, modelos celulares humanos y cortes de tejido cardiaco humano, que restaurar KLF15 puede interrumpir esta reacción en cadena. Aunque aún está en fase preclínica, el sistema CRISPRa compacto y dirigido a cardiomiocitos que se presenta aquí ofrece una hoja de ruta potencial para tratar formas comunes y no genéticas de insuficiencia cardiaca reprogramando la actividad génica en lugar de reescribir el genoma.
Cita: Schoger, E., Kim, R., Bleckwedel, F. et al. Enhancing KLF15 activity in cardiomyocytes: a novel approach to prevent pathological reprogramming and fibrosis via nuclease-deficient dCas9VPR. Sig Transduct Target Ther 11, 76 (2026). https://doi.org/10.1038/s41392-026-02593-9
Palabras clave: insuficiencia cardiaca, KLF15, activación CRISPR, fibrosis cardiaca, AZGP1