Clear Sky Science · en
A high-throughput, quantitative platform using 2D dissociated human cerebral organoids to model neuroinflammation in Alzheimer’s disease
Why infections might matter for memory loss
Alzheimer’s disease is usually described as a slow buildup of sticky proteins in the brain, but growing evidence hints that infections could help set this process in motion. This study explores that idea using tiny lab-grown models of the human brain to ask a simple question: can a common cold-sore virus trigger Alzheimer-like changes in human brain cells, and can an antiviral drug dial those changes back?
Mini-brains on a dish
Instead of working only with animals, the researchers used “cerebral organoids” – clusters of brain-like cells grown from human stem cells. They then gently broke these 3D organoids apart into flat layers of mixed brain cells, including nerve cells, support cells called astrocytes, and immune-like microglia. These two-dimensional cultures, which they call dcOrgs, are easier to infect evenly and to test in high-throughput, meaning many plates and drug conditions can be examined in parallel. This makes the system attractive as a screening tool for new treatments.
A cold-sore virus as a spark
The team infected dcOrgs with herpes simplex virus 1 (HSV-1), the virus responsible for most cold sores and long suspected of contributing to dementia in some people. They compared infected cultures to mock-treated controls, to cultures given an antiviral drug (acyclovir), to cultures exposed to a different virus (influenza A), and to virus that had been inactivated with ultraviolet light. Using automated cell analysis and single-cell sequencing, they confirmed that HSV-1 robustly infected many cell types in the dish, while the inactivated virus and influenza produced very different, milder patterns of change.
Alzheimer-like changes inside and between cells
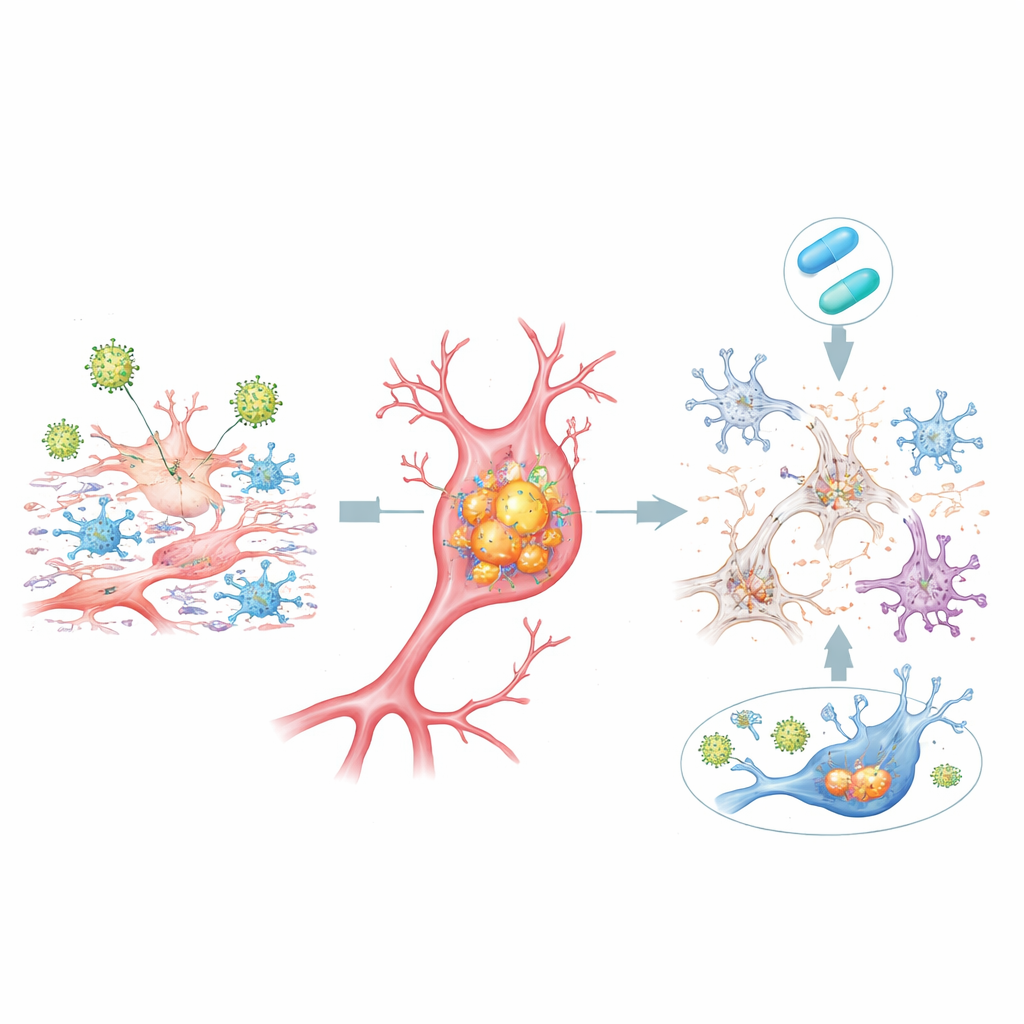
In HSV-1–infected dcOrgs, many cells accumulated high levels of the same protein forms seen in Alzheimer’s brains: clumped beta-amyloid inside cells and multiple “phosphorylated” versions of tau, another key disease protein. These buildups were most tightly linked to cells containing viral proteins and were especially strong in dying cells. At the same time, less of the longer beta-amyloid fragment (Aβ42) was released into the surrounding fluid relative to shorter forms, a shift that mirrors patterns measured in the spinal fluid of patients with Alzheimer’s disease. Cell populations also changed: neuron numbers dropped, while astrocytes and microglia expanded, echoing the neuronal loss and reactive inflammation seen in patient brain tissue.
Gene activity ties the model to human Alzheimer’s
When the researchers examined gene activity across the whole genome, they found that HSV-1 infection in dcOrgs turned on and off many genes previously linked to Alzheimer’s risk in large human genetic studies. These changes were not seen in simpler stem-cell cultures or in influenza-infected dcOrgs, suggesting a specific interplay between HSV-1 and the mixed brain-like environment. Single-cell sequencing revealed that some of the Alzheimer-linked gene shifts came from cells that had been exposed to the virus but contained little or no viral genetic material themselves, hinting that signals from infected neighbors can spread harmful inflammatory programs.
What antiviral treatment can and cannot fix
Adding the antiviral drug acyclovir soon after HSV-1 exposure reduced viral gene expression, dampened many inflammatory responses, lowered the buildup of toxic beta-amyloid and tau inside cells, and partially restored the balance of different cell types. For a substantial fraction of Alzheimer-associated genes, their activity levels moved back toward normal. However, not all changes were reversible: a sizable group of human genes was either left unchanged or even further disturbed by treatment, especially when the drug was less effective at blocking late viral genes. This underscores that while stopping viral replication may help, it may not completely undo the biological cascade once it is underway.
What this means for understanding Alzheimer’s
To a non-specialist, the takeaway is that a human cold-sore virus, acting within a realistic mix of human brain cells, can rapidly produce many hallmarks of Alzheimer’s disease – from protein clumps and dying neurons to genetic patterns already seen in patients. The flat organoid-based system developed here is fast, quantitative, and scalable, making it a powerful testing ground for antiviral drugs and other therapies aimed at calming brain inflammation. Although this does not prove that herpes infections cause Alzheimer’s in every patient, it strengthens the case that, for a subset of people, chronic or reactivated viral infections may be an important piece of the puzzle – and a potential target for prevention.
Citation: Olson, M.N., Barton, N.J., Feng, L. et al. A high-throughput, quantitative platform using 2D dissociated human cerebral organoids to model neuroinflammation in Alzheimer’s disease. npj Dement. 2, 20 (2026). https://doi.org/10.1038/s44400-026-00066-y
Keywords: Alzheimer’s disease, herpes simplex virus, brain organoids, neuroinflammation, antiviral therapy