Clear Sky Science · en
Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design
Why designing better medicines is so hard
Modern drug discovery often hinges on persuading a small molecule to fit into a protein like a key into a lock. But the key must do more than fit: it must form the right set of tiny attractions—such as weak electrical pulls and water‑avoiding patches—so the drug stays bound strongly and specifically. The chemical universe is astronomically large, far beyond what today’s databases contain, so researchers are searching for smarter ways to invent new keys from scratch while preserving these crucial contact patterns.
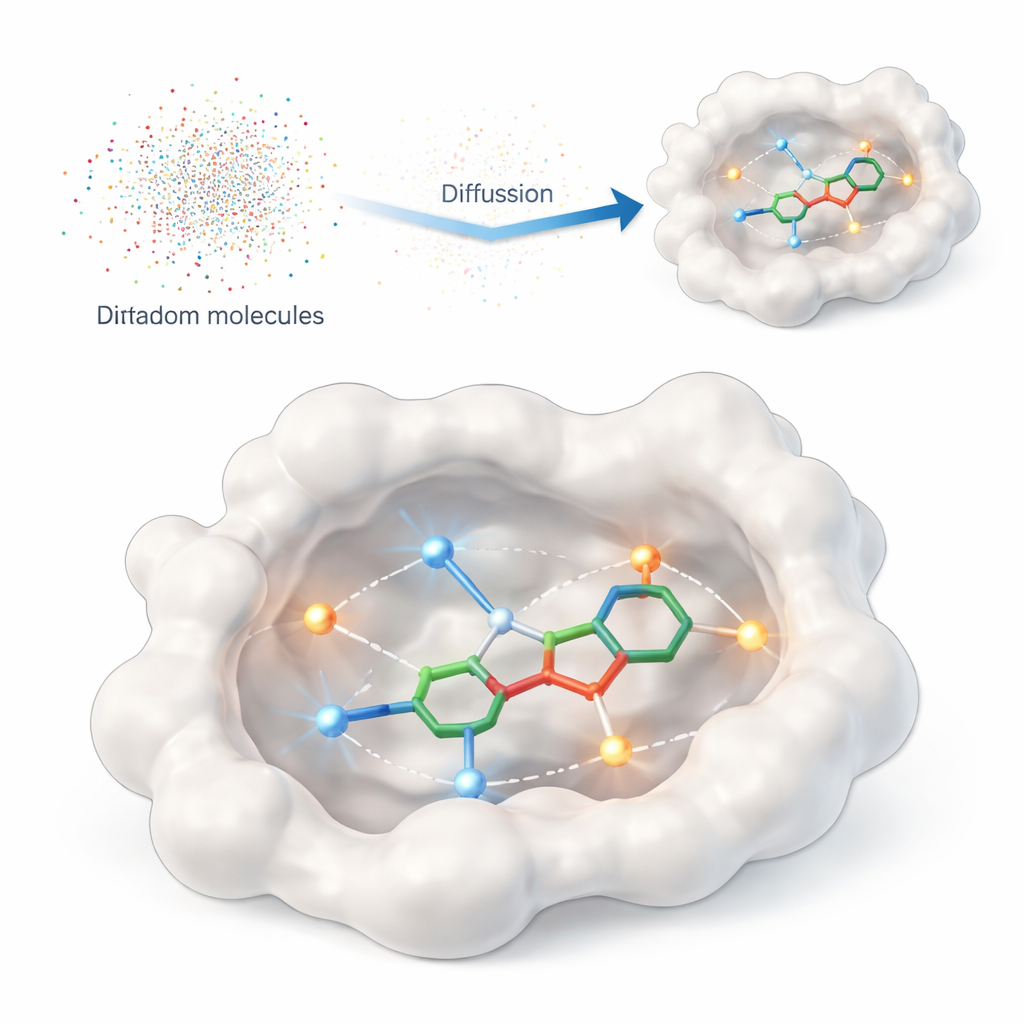
Teaching a computer what really matters
This study introduces DiffPharma, a computational framework that generates three‑dimensional drug‑like molecules directly inside a protein’s binding site. Instead of asking the algorithm to search huge catalogs of existing compounds, DiffPharma creates new ones atom by atom, guided by how they are supposed to interact with the protein. The method is built on a modern class of generative models called diffusion models, which start from random noise and gradually “denoise” it into a structured object—in this case, a 3D molecule nestled in the protein pocket.
Encoding the protein’s handshake
To tell the model what matters at the protein surface, the authors represent key contacts as small “interaction particles” sprinkled along the paths between the protein and a reference molecule. Two common interaction types are emphasized: hydrogen bonds, which act like directional magnets between specific atoms, and hydrophobic contacts, where oily regions cluster together away from water. Separate neural networks learn the geometry and chemistry of each interaction type, as well as the overall shape of the binding pocket, and then a special fusion architecture combines these viewpoints into a single, coherent picture that guides molecule generation.
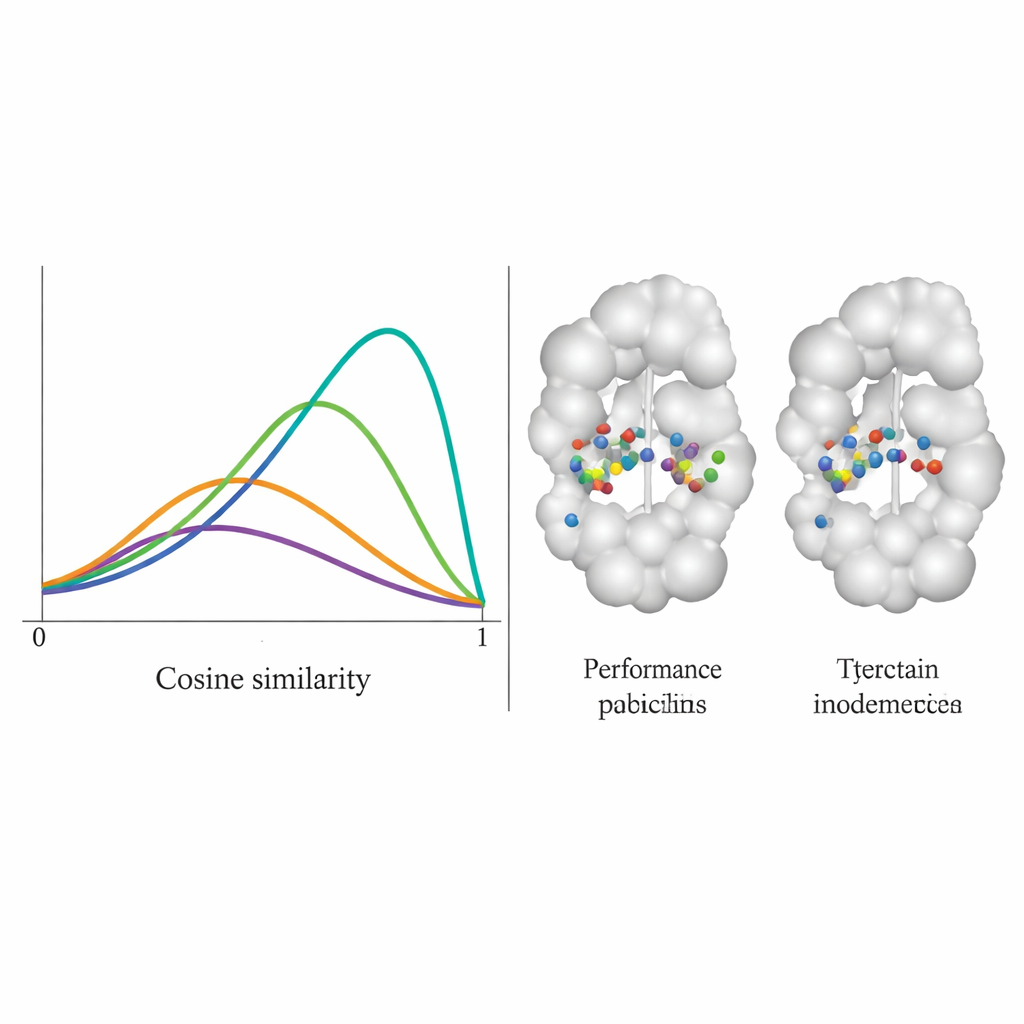
How well does it mimic real binding patterns?
The team tested DiffPharma on 100 different protein–molecule pairs and asked how faithfully new molecules reproduced the original contact patterns, residue by residue. They measured this using a cosine similarity score between 0 and 1, where 1 means perfect agreement. DiffPharma’s distribution peaked around 0.9, meaning that, on average, the same protein residues formed the same types of key interactions as in the reference structures—substantially better than six competing methods. Importantly, the model did this while still producing a variety of molecular shapes, and the generated compounds kept realistic bond lengths, angles, and overall 3D geometry typical of real, stable molecules.
From theory to practical drug leads
Beyond benchmarks, the authors asked whether DiffPharma could design plausible drug candidates for real targets. For two well‑studied enzymes—AKT kinase and a β‑lactamase linked to antibiotic resistance—the method generated molecules that preserved the essential interaction patterns of known ligands yet often used different chemical scaffolds, a desirable form of “scaffold hopping” in medicinal chemistry. In a more demanding case study on the main protease of SARS‑CoV‑2, DiffPharma was steered using specific interaction choices and then examined with molecular dynamics simulations and binding‑energy estimates. Molecules generated under stricter interaction constraints formed more stable complexes and sometimes showed more favorable predicted binding energies than a known reference inhibitor. Notably, the system even rediscovered that reference compound—despite it never appearing in training—purely from the protein structure and interaction instructions.
What this means for future medicines
To a non‑specialist, DiffPharma can be thought of as a smart, 3D‑aware drafting tool for drug molecules: given the shape of a protein pocket and a desired pattern of “handshakes,” it proposes chemically reasonable keys that fit and interact in the right ways. While it does not yet optimize every property a medicine needs, such as solubility or metabolism, the method reliably preserves the crucial contact map at the protein surface and explores new regions of chemical space beyond current catalogs. This interaction‑guided approach may help researchers move more quickly from structural data on disease‑related proteins to diverse, realistic starting points for experimental drug development.
Citation: Sako, M., Yasuo, N. & Sekijima, M. Interaction-constrained 3D molecular generation using a diffusion model enables structure-based pharmacophore modeling for drug design. npj Drug Discov. 3, 8 (2026). https://doi.org/10.1038/s44386-026-00040-x
Keywords: structure-based drug design, molecular generative models, pharmacophore modeling, protein–ligand interactions, SARS-CoV-2 main protease