Clear Sky Science · en
AI-guided competitive docking for virtual screening and compound efficacy prediction
Smarter Searches for New Medicines
Finding new drugs is a bit like looking for a needle in a haystack made of millions of molecules. This study shows how recent advances in artificial intelligence can make that search faster and cheaper by helping scientists predict which molecules are most likely to stick to a disease-related protein and actually work as medicines. Instead of testing one chemical at a time in the lab, the authors use AI models to run virtual contests between molecules and let the winners rise to the top.
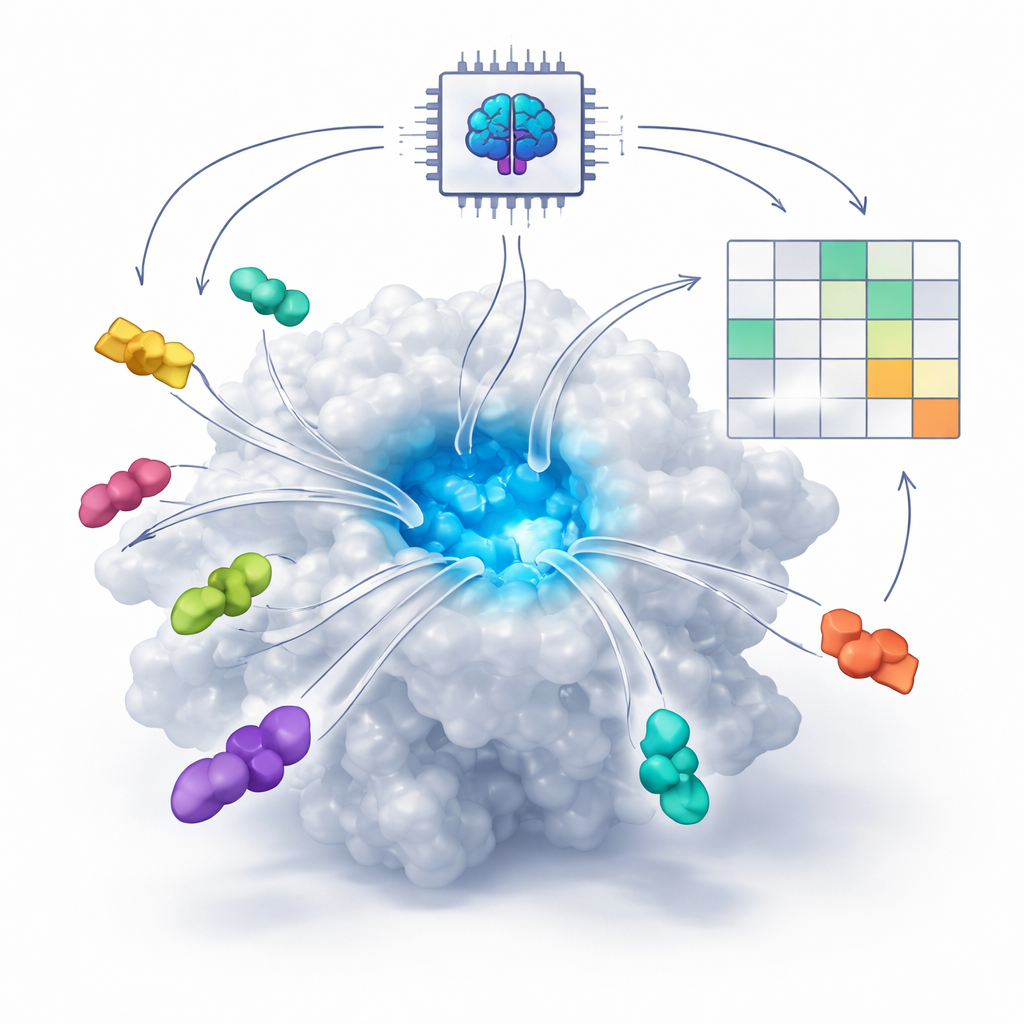
How AI Learns to See Molecular Lock-and-Key Fits
Many modern drugs work by fitting into tiny pockets on proteins, much like a key fits into a lock. Traditionally, computer programs tried to predict this fit using physics equations that estimate forces between atoms. In the last few years, however, new AI systems called diffusion-based co-folding models—such as AlphaFold3 and Boltz—have learned from huge numbers of known protein–molecule structures. These systems can now “imagine” how a protein and a potential drug might fold together in three dimensions, even when no experimental structure exists. The central question the authors tackle is whether these AI tools can do more than just draw plausible pictures—can they also tell good drugs from bad ones?
Real Binders vs. Pretenders
The team first tested 16 well-studied proteins plus a more complex bacterial enzyme called DNA gyrase. For each protein, they asked the AI models to place both known active inhibitors and a set of unrelated “off-target” molecules into the same binding site. Instead of trusting a single prediction, they looked at how consistently the AI placed each molecule across many runs. True inhibitors tended to return to the same spot and orientation over and over again, clustering within a few trillionths of a meter of one another. Inactive molecules wandered more widely and often sat farther from the pocket. This simple idea—pose convergence—turned out to be a strong signal that a compound really fits its protein target.
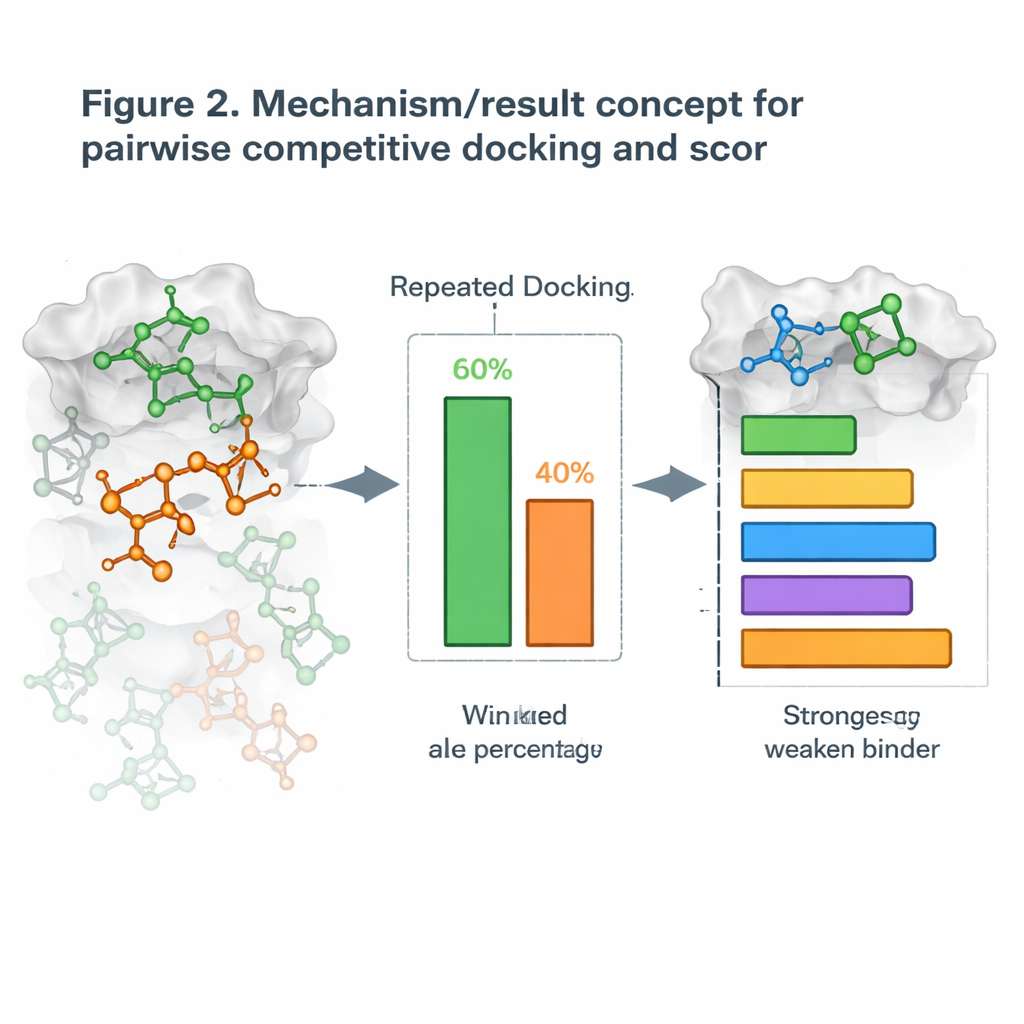
Turning Docking into Head-to-Head Competition
Building on this, the authors introduced a new strategy they call pairwise competitive docking. Rather than docking one molecule at a time, they dock two candidates together with the protein and let them “compete” for the same pocket. After many repeated runs, the molecule that occupies the site more often is declared the winner of that matchup. By running all possible pairings, they build a win–loss table and compute a Competitive Docking Score for each molecule, much like ranking players in a round-robin tournament. When these scores were compared with real-world measurements of how strongly the molecules block their targets, the rankings often matched well, with some protein systems showing near-perfect agreement.
From Virtual Screens to Designing Better Antibiotics
DNA gyrase, an enzyme essential for bacteria, served as a detailed test case. This protein has several drug pockets targeted by different classes of antibiotics, including widely used fluoroquinolones. The AI models could usually place each drug class into its correct pocket, and the competitive docking scores roughly followed their measured potencies. The authors then scaled up to a virtual screen of more than 3,000 approved drugs, asking which molecules best competed for the fluoroquinolone site. Their two-step strategy—first using “all-at-once” competition to pick likely winners, then filtering by how tightly they clustered in the pocket—greatly enriched true fluoroquinolones while discarding weaker candidates. Finally, they used an AI-driven molecule generator to propose new fluoroquinolone-like structures and applied competitive docking to find a handful with even better predicted binding and acceptable drug-like properties.
Promises, Limits, and What It Means for Patients
The study shows that modern AI models can do more than draw plausible protein–drug structures: when run in a competitive framework, they can help rank compounds in a way that often mirrors real experimental data. This does not replace lab work—performance still depends strongly on the particular protein, some pockets are mispredicted, and AI models can fail for very large or unusual molecules. But as these models and their training data improve, approaches like pairwise competitive docking could make early drug discovery far more efficient. For patients, that could eventually translate into faster development of targeted medicines, including new antibiotics that keep pace with resistant bacteria.
Citation: Mirgaux, M., Barcelli, V., Chua, A.C.Y. et al. AI-guided competitive docking for virtual screening and compound efficacy prediction. npj Drug Discov. 3, 6 (2026). https://doi.org/10.1038/s44386-026-00039-4
Keywords: AI drug discovery, virtual screening, molecular docking, protein-ligand binding, antibiotic design