Clear Sky Science · en
Energy-efficient scientific computing using chemical reservoirs
Why turning chemistry into computing matters
Modern supercomputers burn through enormous amounts of electricity to simulate climate, design new drugs, or train artificial intelligence. As we approach the physical limits of traditional chips, squeezing out more performance per watt is getting harder and more expensive. This paper explores a radically different path: using real chemical reactions as the engine of scientific computing. By treating molecules and their interactions as the moving parts of a computer, the authors outline how future machines could solve complex equations with far less energy than today’s digital hardware.
From living cells to chemical calculators
Living cells are master problem-solvers. They constantly juggle thousands of reactions to adapt, grow, and survive, all while using remarkably little energy. At the heart of this behavior are chemical reaction networks—interconnected reactions whose rates and concentrations change over time. These networks can be described by ordinary differential equations, the same mathematical language used to model everything from epidemics to turbulent flows. The insight behind this work is that if chemistry already follows these equations, we might harness it directly to carry out the calculations that scientists now perform on silicon chips.
How equations become reaction networks
The authors introduce ChemComp, a software framework that takes a system of differential equations and systematically converts it into an abstract network of reactions. ChemComp uses modern compiler technology to break down a mathematical problem into patterns that can be represented by idealized reactions, then organizes them into a network with well-defined species, connections, and rates. These abstract reactions do not yet correspond to real molecules, but they form a blueprint for a chemical computer. The framework can then search biochemical reaction databases to find real reaction motifs that behave similarly, favoring options that are practical, safe, and potentially energy-efficient in a lab setting.
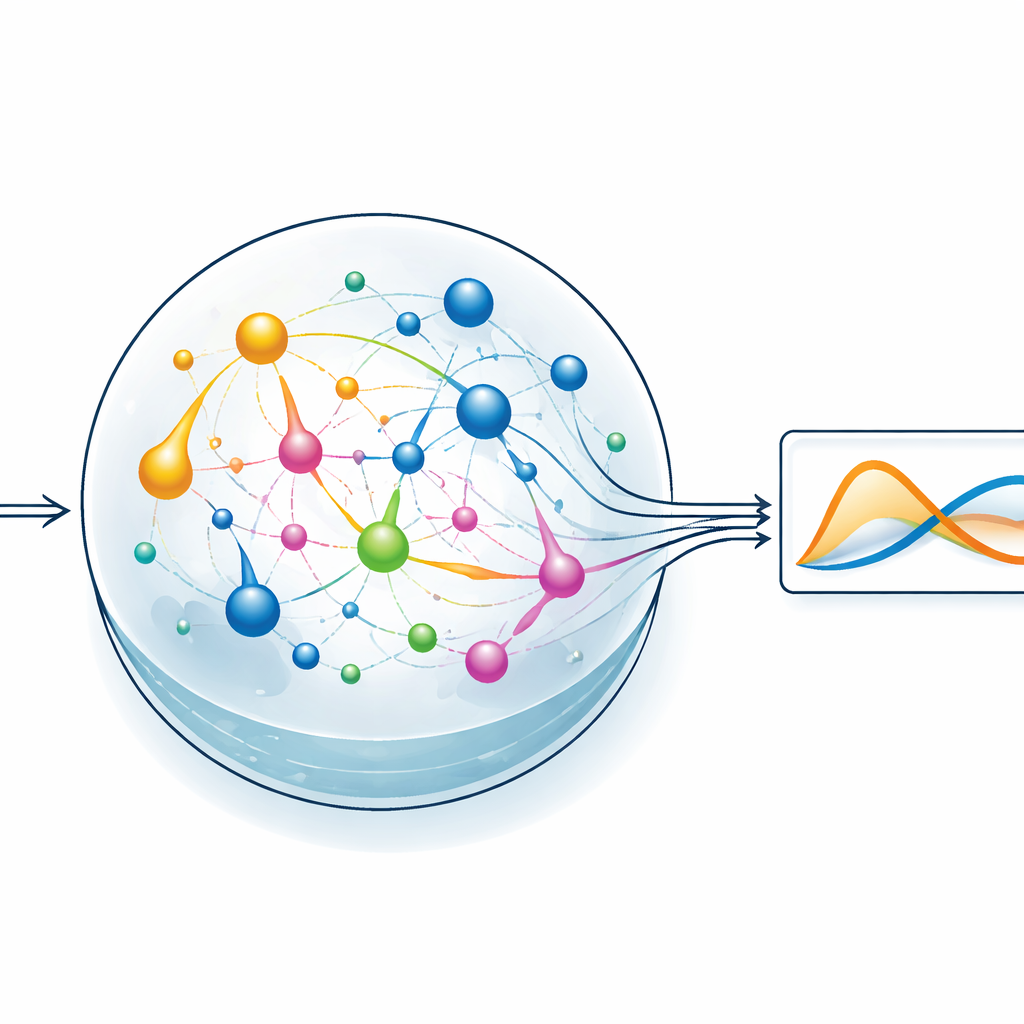
Letting a chemical reservoir do the hard work
To test the idea, the team focuses on a style of machine learning called reservoir computing. Here, a fixed, dynamic system transforms an input signal into a rich, tangled pattern of internal activity, and only a simple readout layer is trained to produce the desired output. In ChemComp’s version, the reservoir is a set of reactions in a well-stirred vessel; the changing concentrations of chemicals form the internal states. The authors compile a classic two-variable system known as the Sel’kov–Schnakenberg model—originally used to study oscillations in metabolism—into candidate reaction networks. They then simulate how these networks respond over time when driven by flows of chemicals in and out of the vessel, and use basic linear regression to combine the concentration traces into an approximation of the target solution.
Testing simple and richer chemical networks
The researchers compare two candidate reservoirs: one with just two chemical species and two reactions, and another with five species and five reactions. Both networks are given suitable starting concentrations and flow rates, then simulated as they run. Even the smaller system can roughly reproduce the oscillating behavior of the target equations, but the larger network does noticeably better, reducing the error during both training and testing. By scanning over different initial concentrations and reaction rate constants, the authors map out regions where the chemical system most closely matches the desired dynamics. Each reaction effectively acts like a basis function in a curve-fitting problem: the more varied reactions available, the easier it becomes to approximate complex behavior, at the cost of added system complexity.
Path toward low-energy computing in the lab
Beyond simulations, the paper looks ahead to practical devices. It discusses how reaction choice must balance energy use, controllability with enzymes or catalysts, and the ability to measure key species in real time, for example with optical or electrochemical methods. The authors suggest that future microfluidic platforms could host carefully chosen reaction networks, with spatial control of inputs and built-in sensing. While many engineering challenges remain—from mapping equations to real chemistry to handling noise and measurement limits—the study shows that modest reaction systems can already emulate the solutions of coupled differential equations. For a lay reader, the core message is that chemistry itself can act as an analog computer, opening a path to scientific calculations that ride on the energy-efficient processes nature has been perfecting for billions of years.
Citation: Johnson, C.G.M., Bohm Agostini, N., Cannon, W.R. et al. Energy-efficient scientific computing using chemical reservoirs. npj Unconv. Comput. 3, 17 (2026). https://doi.org/10.1038/s44335-026-00053-9
Keywords: chemical computing, energy-efficient computing, reservoir computing, chemical reaction networks, ordinary differential equations