Clear Sky Science · en
An end-to-end framework for reactivity in heterogeneous catalysis
Why faster catalyst design matters
Modern society leans on catalysts to make fuels, plastics, fertilizers and countless everyday products. Yet finding better catalysts is often like searching for a needle in a haystack, because each material can promote thousands of possible microscopic reactions at once. This article introduces CARE, a new computational framework that uses smart rules and machine learning to map out and simulate these tangled reaction webs much faster and more completely than before. By doing so, it promises to guide cleaner energy technologies and more efficient chemical processes while slashing computing costs.
Untangling crowded reaction pathways
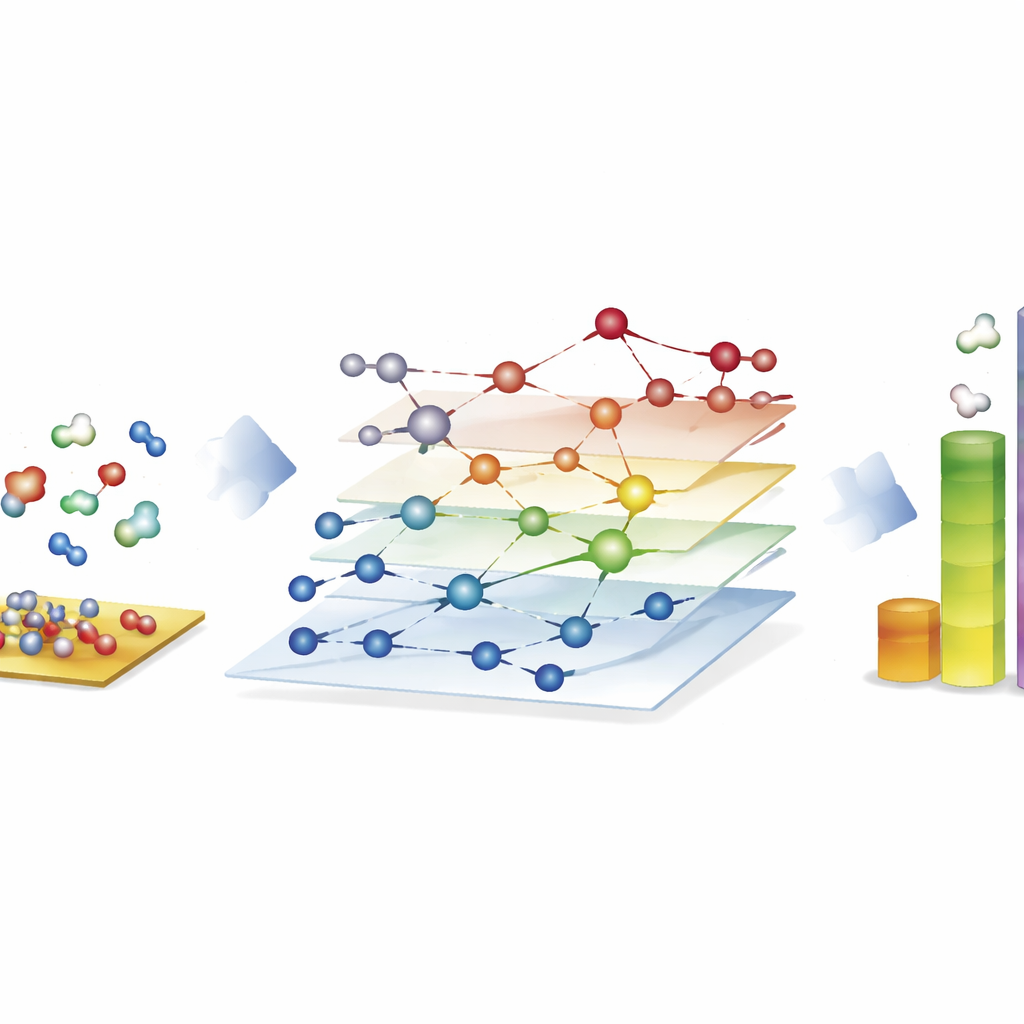
On the surface of a solid catalyst, incoming molecules do not simply follow a single neat route from reactant to product. Instead they travel through a maze of short-lived intermediates and competing pathways. Traditional computer methods rely on human intuition to pick a limited set of possible steps and then use quantum calculations to evaluate their energies. This works for small networks but quickly breaks down as systems grow more complex, overlooking rare routes that may govern long-term activity, deactivation or selectivity. CARE tackles this challenge by automatically constructing very large reaction networks from simple building rules, ensuring that all plausible bond-breaking and bond-making events between carbon, hydrogen and oxygen are included, even those chemists might normally discard.
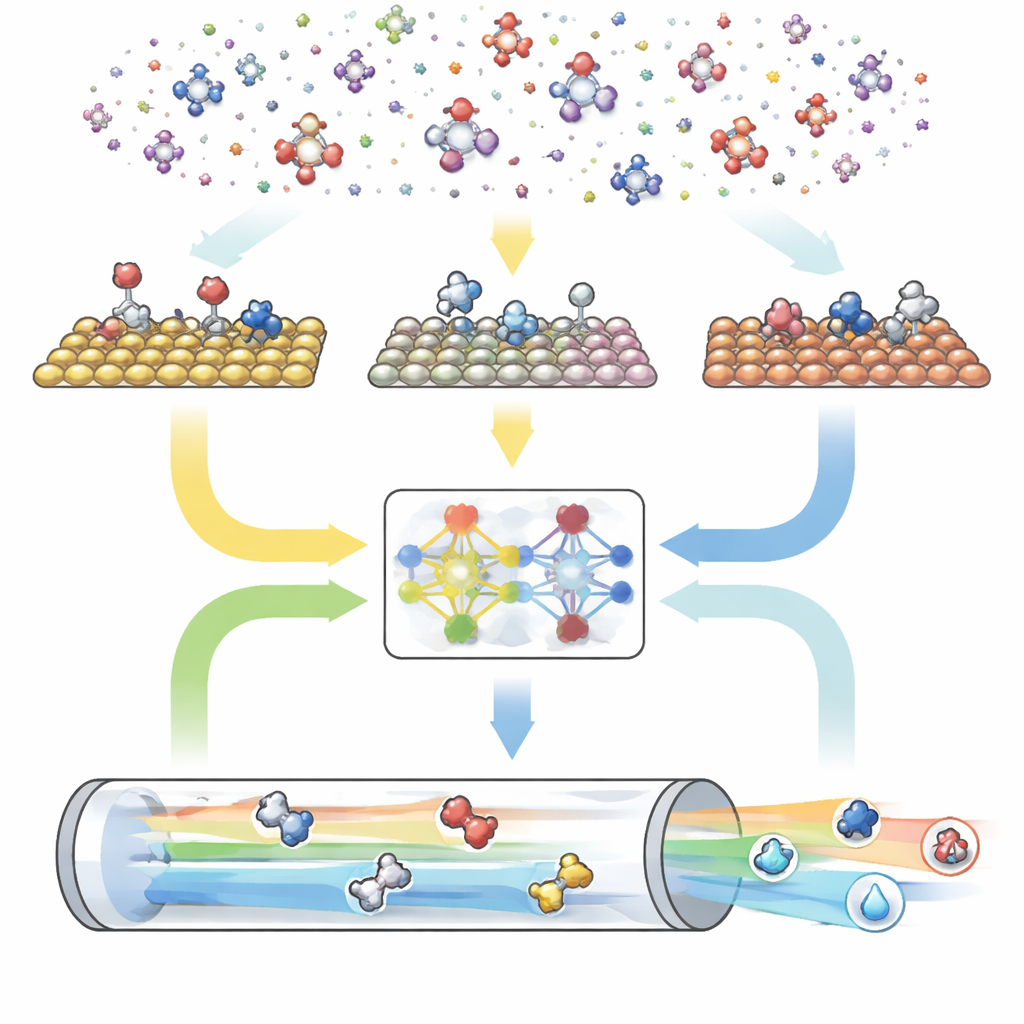
A three-part digital engine for reactions
CARE is built as an end-to-end pipeline with three main modules. First, a rule-based generator defines the “chemical space” by choosing the maximum number of carbon and oxygen atoms and then applying simple templates to create all matching molecules and their surface-bound forms. Second, an energy evaluation module calls modern machine learning models—especially a graph neural network called GAME-Net-UQ—to estimate the energies of intermediates and transition states on many metal surfaces. This model treats each structure as a network of atoms and bonds, returns both an energy and an uncertainty, and is accurate to within a few tenths of an electronvolt while remaining lightweight and fast. Third, a microkinetic solver uses these energies to compute how all reactions proceed together under realistic conditions of temperature, pressure, voltage and pH, predicting overall reaction rates, surface coverages and product selectivity.
Real-world tests: fuel molecules and climate chemistry
To show that CARE is not just a theoretical exercise, the authors apply it to three industrially relevant problems of increasing difficulty. For methanol decomposition—a reaction important for hydrogen storage—they generate a modest network and evaluate it across many metal catalysts and crystal faces. CARE reproduces the familiar “volcano” trend in activity and correctly identifies ruthenium as one of the best performers, in line with experiments, but at a tiny fraction of the computing time required for full quantum calculations. Next, they move to the electrochemical conversion of carbon dioxide on copper, focusing on how three-carbon products such as 1-propanol and propylene arise. By including special steps that account for protons, electrons and solution conditions, CARE captures how pH and applied voltage shift pathways and correctly predicts that 1-propanol is favored over propylene, echoing detailed prior studies.
Exploring huge reaction webs for synthetic fuels
The most striking demonstration comes from the Fischer–Tropsch process, which turns mixtures of carbon monoxide and hydrogen into long-chain hydrocarbons for fuels and chemicals. Here, the authors construct networks with nearly 40,000 surface species and about 370,000 elementary reactions—far beyond what traditional quantum-based studies can fully explore. Using CARE, they evaluate all intermediates and key reaction barriers on cobalt, iron, nickel and ruthenium surfaces in only a few hours on standard hardware, a speed-up of about a million-fold compared with direct quantum calculations. Microkinetic simulations on these networks reproduce known trends: cobalt and iron preferentially form longer hydrocarbon chains, iron produces more carbon dioxide through side reactions, and nickel tends toward stronger hydrogenation. Although some details, such as methane yields, remain imperfect, the framework reveals which bond-forming steps dominate chain growth and highlights where models still need refinement.
What this means for future catalysts
For non-specialists, the key message is that CARE provides a practical way to explore enormous reaction spaces on catalytic surfaces that were previously out of reach. By automating network generation, plugging in fast machine-learning “surrogate” models for quantum energies and solving the resulting kinetics efficiently, it can rank candidate catalysts, identify promising operating conditions and uncover unexpected pathways with far less human bias and computational expense. While the authors note remaining challenges—such as better handling of crowded surfaces, solvent effects and even larger networks—the work points toward a future where computers can rapidly screen complex reactions, from carbon dioxide reduction to plastic recycling and biomass upgrading, guiding experiments toward the most promising ideas rather than leaving discovery to trial and error.
Citation: Morandi, S., Loveday, O., Renningholtz, T. et al. An end-to-end framework for reactivity in heterogeneous catalysis. Nat Chem Eng 3, 169–180 (2026). https://doi.org/10.1038/s44286-026-00361-8
Keywords: heterogeneous catalysis, reaction networks, machine learning, microkinetic modeling, Fischer–Tropsch synthesis