Clear Sky Science · en
Hematopoietic expression of cIAP2 drives inflammation and heart failure after myocardial infarction
Why calming post-heart-attack inflammation matters
Surviving a heart attack is only the beginning of the story. In the days and weeks that follow, the body’s immune system rushes in to clean up damaged tissue and start repairs. But if this inflammatory response burns too hot or lasts too long, it can turn helpful healing into lasting heart damage and heart failure. This study uncovers a key molecular switch inside blood-forming immune cells that keeps this inflammatory fire going—and shows that flipping that switch off can protect the heart in experimental models.
A hidden culprit inside immune cells
The researchers focused on a protein called cIAP2, best known for helping cancer cells avoid death. Using blood samples from patients with acute heart problems, they found that cIAP2 levels were higher in people with recent heart attacks and ischemic heart failure than in healthy individuals or patients with stable coronary disease. Heart tissue from humans and mice showed the same pattern: cIAP2 surged shortly after a heart attack, while its close relative cIAP1 did not. Mining existing gene-expression datasets, the team saw that cIAP2 levels rose in lockstep with genes linked to aggressive, myeloid-type inflammatory cells, hinting that cIAP2 might be amplifying the post-attack immune response rather than simply reflecting damage.
Less cIAP2, less damage to the heart
To test causality, the team compared normal mice with mice genetically engineered to lack cIAP2. After an experimental heart attack, animals without cIAP2 had smaller scars, better pumping function, and less fluid buildup in the lungs, all signs of healthier hearts. These benefits appeared in both males and females. Microscopy showed fewer dying heart muscle cells in critical border regions, and molecular analyses revealed lower levels of stress and remodeling markers weeks later. In contrast, knocking out cIAP1 did not offer the same protection and could even worsen outcomes in some settings, pointing to a unique, harmful role for cIAP2 in this context.
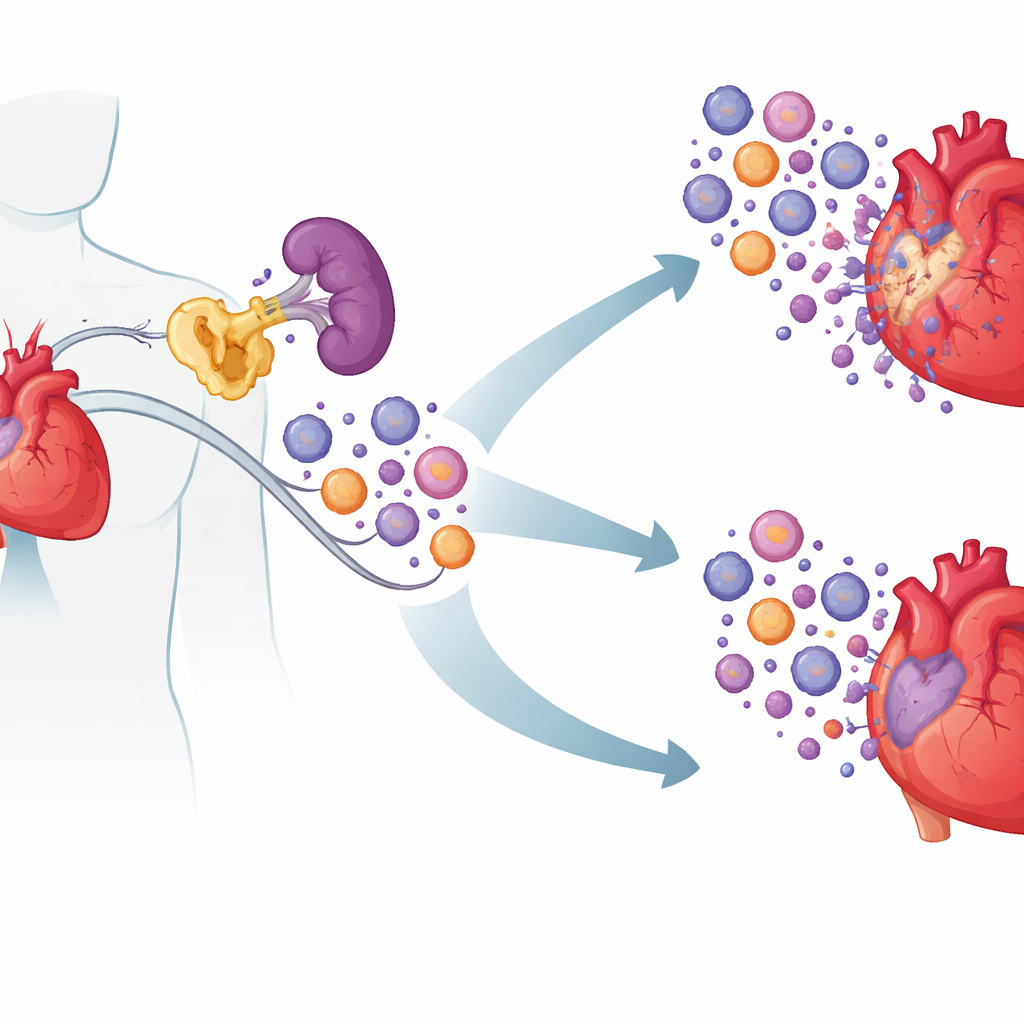
The spleen’s role as an inflammatory reservoir
The key turned out to be where cIAP2 was acting. By swapping bone marrow between normal and cIAP2-deficient mice, the researchers showed that cIAP2 inside blood-forming (hematopoietic) cells drove much of the injury. When immune cells lacked cIAP2 but the rest of the body was normal, hearts were better protected; the opposite swap made damage worse. Zooming in on immune organs, they found that after a heart attack, the spleen acted as a reservoir churning out myeloid cells—neutrophils, inflammatory monocytes, and dendritic cells—that then streamed into the heart. In mice missing cIAP2, these splenic myeloid cells were fewer and more prone to die, while lymphocytes were largely unaffected. Signals linked to inflammatory pathways were dampened, suggesting that cIAP2 normally helps myeloid cells survive and keep responding to danger signals.
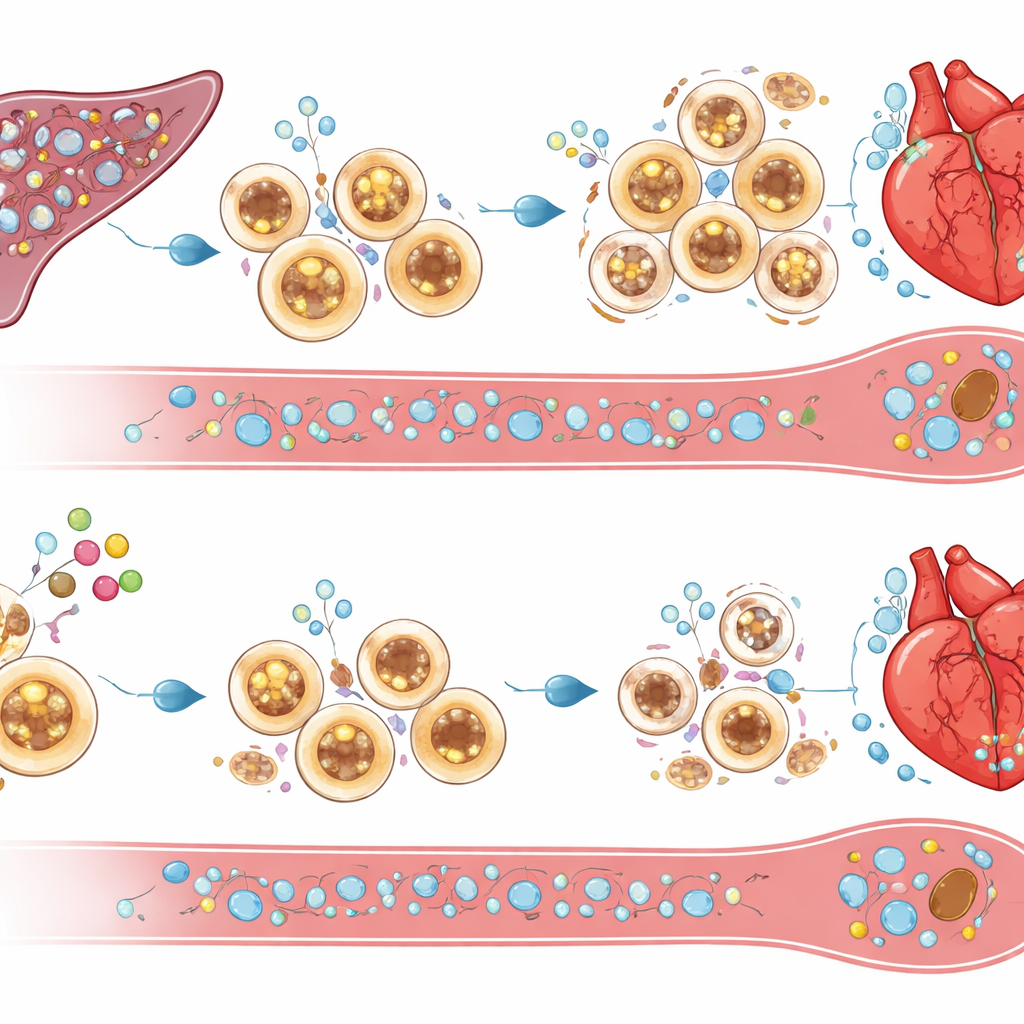
Turning survival signals into self-limited cleanup
What kills excess inflammatory cells when cIAP2 is missing? The study points to death-inducing molecules such as TRAIL and its receptor DR5, along with TNF-related signals, which were upregulated in the spleens and bone marrow of cIAP2-deficient mice after heart attack. Blocking TRAIL experimentally rescued splenic cells from death, restored heavy immune-cell infiltration of the heart, and erased the functional benefits of losing cIAP2. This suggests that cIAP2 normally shields myeloid cells from these death cues, allowing them to accumulate and prolong inflammation. Without cIAP2, the same cues prune the splenic reservoir, trimming back the supply of aggressive cells that would otherwise flood the injured heart.
Drugging the switch for future therapies
Importantly, the team showed that this pathway can be targeted with an existing class of small molecules known as Smac mimetics, currently under study for cancer. Using the compound LCL161, they selectively triggered the breakdown of cIAP proteins in splenic immune cells shortly after a heart attack, without depleting protective proteins in heart tissue itself. Treated mice had fewer circulating inflammatory cells, smaller scars, better heart function, and improved survival compared with untreated animals. A single low-dose treatment given one day after heart attack was enough to induce controlled death of splenic myeloid cells, boost TRAIL levels locally, and reduce heart inflammation, while total immune cell numbers recovered by four weeks. Together, these findings position cIAP2 as a central survival factor for inflammatory cells after heart injury and suggest that short-term, targeted inhibition of cIAP2 could offer a new immunotherapy-style approach to prevent heart failure following a heart attack.
Citation: Smyth, D., Zhang, L., Al-Khalaf, M. et al. Hematopoietic expression of cIAP2 drives inflammation and heart failure after myocardial infarction. Nat Cardiovasc Res 5, 246–261 (2026). https://doi.org/10.1038/s44161-026-00782-x
Keywords: myocardial infarction, inflammation, immune cells, heart failure, Smac mimetic