Clear Sky Science · en
Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation
Why this heart gene matters to families
Many sudden heart problems in young and otherwise healthy people are not random—they are written, at least in part, in their DNA. This article explores a key heart “glue” protein called desmoglein‑2 and shows how small changes in its gene can weaken the heart’s muscle, disrupt its electrical rhythm, and raise the risk of dangerous events. By combining big genetic databases with a carefully followed Italian patient group, the researchers offer clearer answers for families who wonder what a test result for this gene really means.
The heart’s mechanical glue
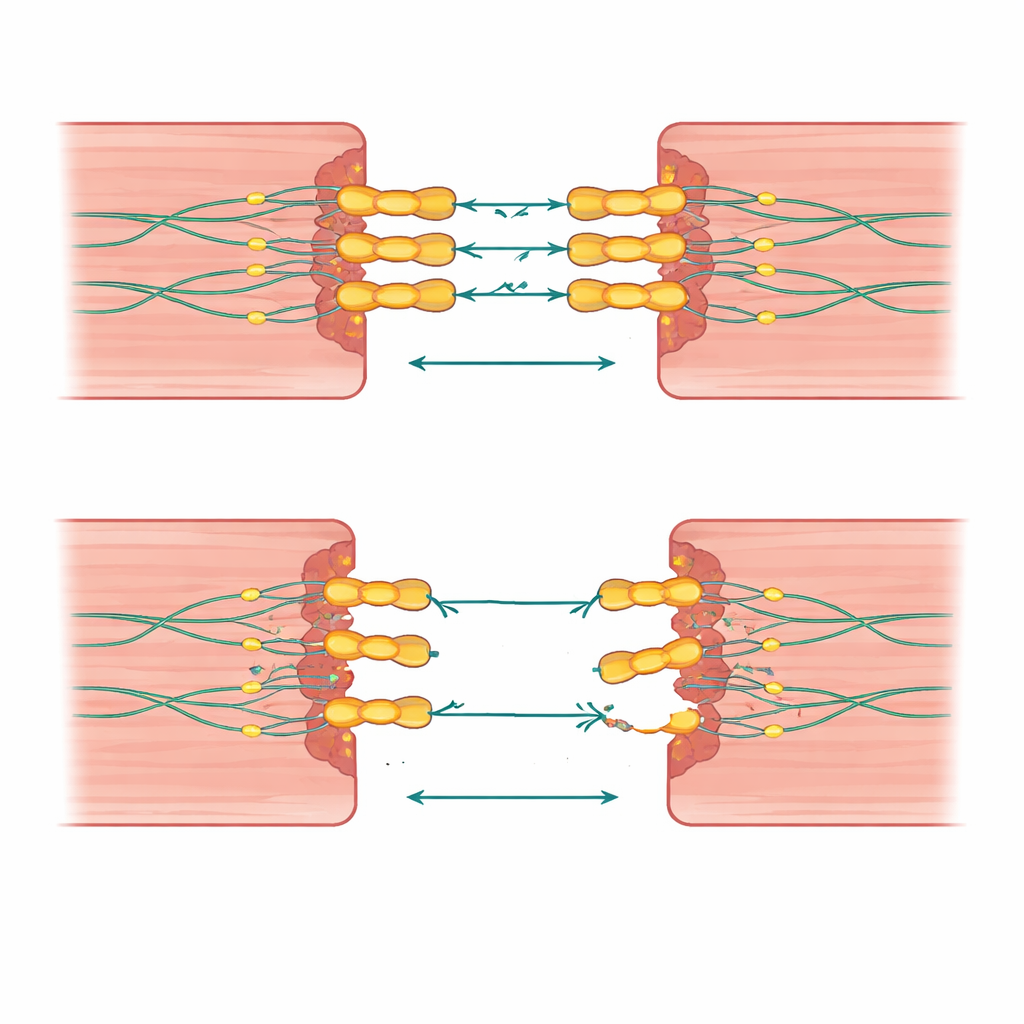
Heart muscle cells must cling tightly to each other while beating millions of times in a lifetime. Desmoglein‑2 is part of a microscopic rivet-like structure that locks neighboring cells together so they can pull as a team. The authors explain how this protein spans from the outside of the cell, where it grips a matching partner on the next cell, to the inside, where it hooks into a supporting framework. Because desmoglein‑2 is the only member of its family present in heart cells, any serious damage to it cannot be backed up by a substitute, making the heart especially vulnerable.
Sorting meaningful gene changes from background noise
Modern sequencing finds thousands of differences in the desmoglein‑2 gene across the population, but most do not cause disease. The team systematically reviewed 115 published studies and drew on two large public databases that together listed more than 5,000 variants. Using widely accepted medical genetics rules, they re‑classified each change by how likely it is to be harmful. They found that truly damaging variants cluster in specific regions of the protein—especially the outer segments that need calcium to form a stiff bridge between cells, a short stretch that must be cut for the protein to mature, and the inner region that latches onto another key heart protein. Many other changes remained “uncertain,” but a subset showed strong hints of being important and were flagged for closer follow‑up.
What the Italian patient group reveals
To see how these genetic patterns play out in real people, the researchers studied 95 individuals in Italy who carried desmoglein‑2 variants and were evaluated in depth with scans, heart rhythm tests, and long‑term follow‑up. About half met strict criteria for arrhythmogenic cardiomyopathy, a condition in which parts of the heart muscle are slowly replaced by scar and fat, setting the stage for dangerous rhythm disturbances. Among relatives who carried a variant, only about four in ten actually showed signs of disease, underlining that a positive gene test does not guarantee illness but does signal the need for careful monitoring. Those with clear disease had a noticeable burden of serious rhythm events, while transplants and deaths were less frequent but still present.
When one hit is not enough
A striking insight from this work is that the number and combination of gene changes matter. People who inherited two faulty copies of desmoglein‑2, or one desmoglein‑2 variant plus a change in a related heart‑glue gene, tended to fall ill younger and show more widespread damage to both sides of the heart. Some families carried large deletions or duplications that removed or doubled not only desmoglein‑2 but also neighboring genes, again tying these alterations to aggressive disease and clusters of sudden deaths. In contrast, many relatives with only a single change had mild or no symptoms, suggesting that background genes and life factors such as exercise may tip the balance between silent risk and overt disease.
From protein shape to patient risk
To link the DNA code to physical effects, the team used advanced 3D protein models to see how specific substitutions might loosen the desmoglein‑2 scaffold. Changes that distorted calcium‑binding loops or broke key attachment points were predicted to destabilize the protein and weaken cell‑to‑cell adhesion. These structural clues were folded back into the classification system, helping to push some borderline variants toward being considered more likely harmful or more likely harmless. This bridge between molecular modeling and clinical data moves genetic testing beyond simple code reading toward a more functional understanding.
What this means for patients and families
For families touched by arrhythmogenic cardiomyopathy, this study offers both caution and guidance. It shows that not every desmoglein‑2 variant is a sentence to severe heart disease, but that certain patterns—especially multiple hits or changes in critical regions of the protein—are linked to earlier and more serious problems. The authors argue that people carrying these variants should not be dismissed as “healthy until proven otherwise,” but instead followed over a lifetime with tailored rhythm checks and imaging. Their integrative approach—blending big‑data genetics, detailed family studies, and protein structure—points the way toward more precise risk estimates and more confident counseling when a desmoglein‑2 change appears on a genetic test.
Citation: Pinci, S., Celeghin, R., Martini, M. et al. Integrative genomic and literature assessment of desmoglein 2-related arrhythmogenic cardiomyopathy with Italian cohort validation. Commun Med 6, 145 (2026). https://doi.org/10.1038/s43856-026-01416-w
Keywords: arrhythmogenic cardiomyopathy, desmoglein-2, inherited heart disease, genetic risk, sudden cardiac death