Clear Sky Science · en
A highly resolved integrated single-cell atlas of HPV-negative head and neck cancer
Why this cancer map matters
Head and neck cancers that are not caused by human papillomavirus (HPV) are common, often aggressive, and notoriously unpredictable: two patients with similar-looking tumors can respond very differently to the same treatment. This study set out to understand why by zooming in to the level of individual cells. The authors merged single-cell data from more than 230,000 cells across 54 patients into one detailed “atlas” of HPV-negative head and neck cancer. This atlas reveals which cells are present in tumors, how they interact, and how they may shape treatment response and patient outcome.
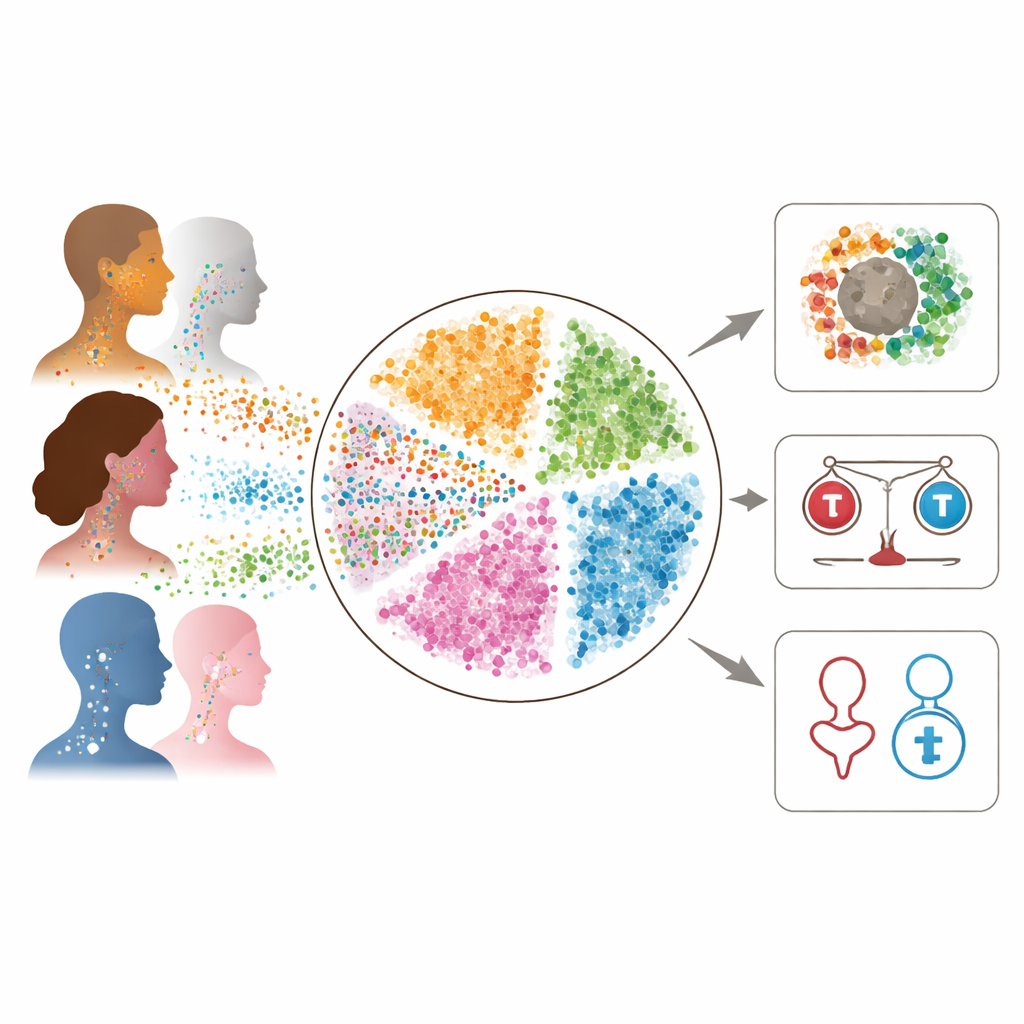
Building a high‑resolution cancer atlas
The researchers combined six previously published single-cell RNA sequencing datasets from patients whose cancers arose mostly in the mouth and voice box and were linked to smoking or alcohol use rather than HPV infection. Each dataset had been generated with slightly different methods, so the team applied careful computational steps to clean the data, remove poor-quality cells, and harmonize cell labels. They then used advanced algorithms to integrate the datasets into a single coherent map where cells clustered by biological type (such as tumor cells, immune cells, or blood vessel cells) rather than by study of origin. This integration created a powerful resource: a shared reference that captures both the diversity and common patterns of HPV-negative head and neck tumors.
Who lives inside these tumors?
Within the immune “neighborhood” of the tumors, the atlas distinguished many subtypes of T cells, B cells, plasma cells, macrophages, monocytes, dendritic cells, and neutrophils. By scoring cells with known gene programs, the authors traced how tumor‑killing CD8 T cells and natural killer cells can shift along a continuum from highly cytotoxic to dysfunctional, exhausted states. They found that early-stage tumors (stage T1) are enriched for more effective, cytotoxic CD8 T cells when considered across the entire atlas, an association that was too subtle to detect in any single study alone. The team also organized immune clusters into a “family tree,” showing how related subtypes group together and which combinations of cell types tend to rise and fall together across patients, patterns that mirror good or poor prognosis in earlier work.
Hidden players: special myeloid cells and fibroblasts
A major payoff of the larger dataset was the ability to resolve fine-grained subpopulations in the tumor microenvironment. Among myeloid cells, the atlas recaptured two macrophage states previously linked to cancer outcome but also highlighted a distinct IL1B‑rich population that had been inconsistently labeled across earlier studies. These cells produce inflammatory and immunosuppressive molecules, and show unique signaling patterns involving tumor necrosis factor, interleukin‑1β, and a matrix protein called thrombospondin, all of which have been tied to tumor growth, drug resistance, or blood vessel changes. In the stromal compartment, the authors dissected cancer‑associated fibroblasts into multiple groups, including two separate inflammatory fibroblast types: one centered on the chemokine CXCL8 and another on CXCL12. They showed that the CXCL8‑rich fibroblasts preferentially signal to blood vessel cells via a receptor called ACKR1, a route that other studies suggest could foster new vessel growth and worse outcomes.
Tumor edge cells and sex-linked differences
The epithelial compartment — the cancer’s main body — also showed striking structure. Using DNA copy-number patterns, the team separated normal from malignant epithelial cells and then ordered them along a differentiation and “plasticity” scale. One cluster, labeled Epi1, combined stem‑like features, partial epithelial‑to‑mesenchymal transition (a program linked to invasion and therapy resistance), and high developmental potential. By comparing with a spatial dataset from other patients, the authors found that Epi1 cells align with tumor “leading-edge” regions at the invasive front, where tumor cells meet and interact with supportive stromal cells. Communication analyses revealed that these edge cells both send and receive intensive extracellular matrix and growth-factor signals — particularly TGF‑β — from fibroblasts and blood vessel cells. Finally, by leveraging the large cohort, the study uncovered sex‑associated shifts in cell composition: male patients had higher proportions of certain macrophages and proliferating and CD8 T cells, and more of the aggressive Epi1 and another epithelial cluster, while female patients had relatively more plasma cells, monocytes, and natural killer cells.
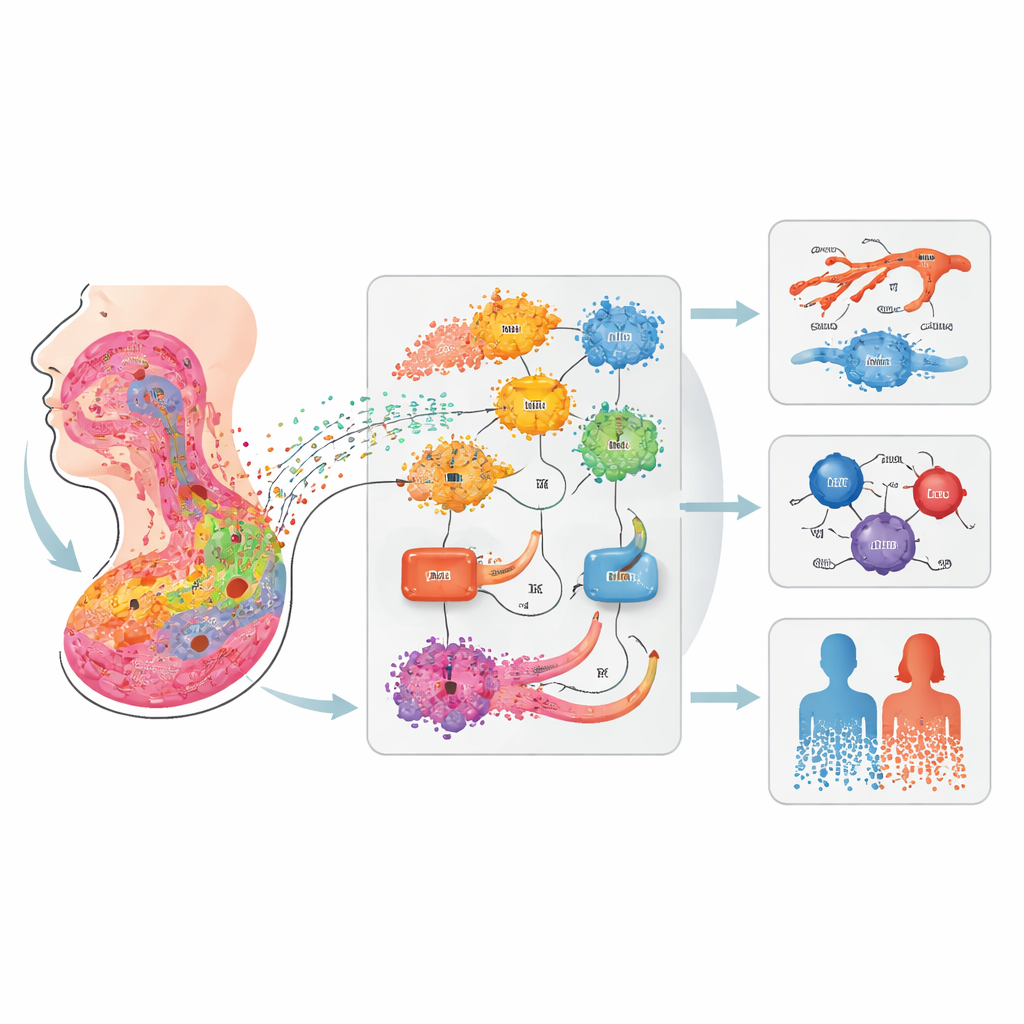
What this means for patients and future research
Taken together, this work turns scattered single‑patient datasets into a unified, public atlas of HPV‑negative head and neck cancer at single‑cell resolution. For non-specialists, the key message is that a tumor is not just a mass of identical cancer cells: it is an ecosystem in which specific immune cells, fibroblasts, and invasive edge cells can either restrain or fuel disease and influence who benefits from immunotherapy or other treatments. By clarifying the identities and interactions of cell populations such as IL1B‑positive myeloid cells, CXCL8‑producing fibroblasts, and stem‑like edge epithelial cells, the atlas points to concrete cellular targets and signaling pathways that could be tested for new drugs or combination therapies. Just as importantly, it provides a common language and reference map so that future studies can more easily compare findings, explore sex differences, and link molecular patterns to clinical outcomes in a more precise and personalized way.
Citation: Kroehling, L., Chen, A., Spinella, A. et al. A highly resolved integrated single-cell atlas of HPV-negative head and neck cancer. Commun Med 6, 138 (2026). https://doi.org/10.1038/s43856-026-01401-3
Keywords: head and neck squamous cell carcinoma, single-cell RNA sequencing, tumor microenvironment, cancer-associated fibroblasts, tumor immune landscape