Clear Sky Science · en
Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles
Why protein motion matters
Proteins in our cells are not rigid sculptures; they behave more like tiny, flexible machines that constantly shift their shapes. These shape changes can control how enzymes catalyze reactions, how receptors respond to drugs, and how signals flow through cells. Yet most familiar images of proteins show just one “snapshot” structure, missing the rich ensemble of forms that actually exist. This paper introduces Mac-Diff, an artificial intelligence method that can quickly generate many realistic shapes for a given protein, helping scientists see not just what a protein looks like, but how it breathes and moves.
From single snapshots to moving ensembles
For decades, researchers have relied on painstaking experiments or long-running molecular dynamics simulations to explore protein motion, both of which can be slow and expensive. Breakthrough tools like AlphaFold2 now predict a protein’s most likely 3D structure directly from its amino-acid sequence, but usually return just one or a few preferred shapes. Many proteins, especially those involved in signaling and allosteric regulation, naturally occupy multiple, loosely defined states. The authors argue that to understand how such proteins really work—and to design drugs that bind to less obvious, transient forms—we need a way to generate entire ensembles of plausible conformations, not just one best guess.
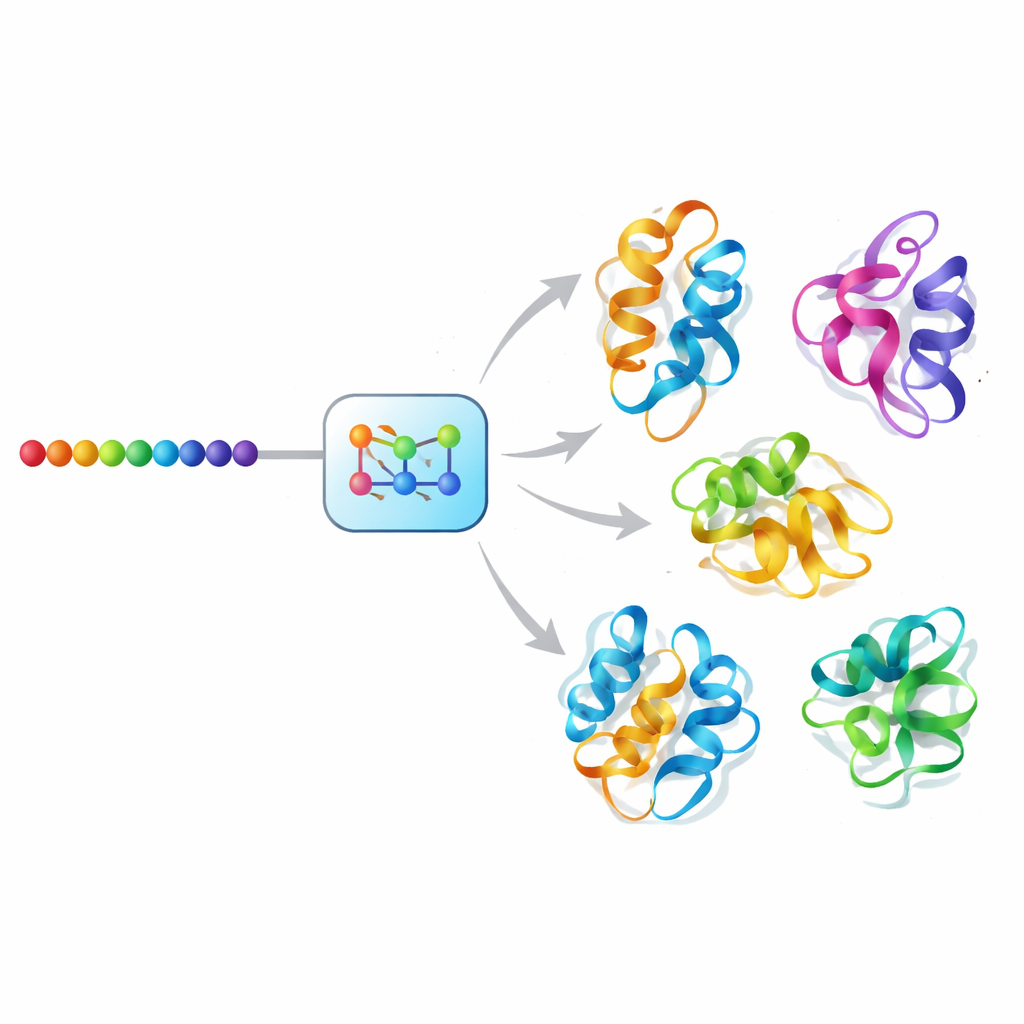
An AI "diffusion" approach to protein motion
Mac-Diff tackles this challenge using a diffusion-style generative model, a class of AI that has powered recent leaps in image synthesis. Instead of denoising photographs, Mac-Diff denoises abstract geometric descriptions of protein backbones. The model represents a protein as a grid of pairwise relationships between its residues—distances and angles that are insensitive to how the whole molecule is rotated or translated. In a forward step, the system gradually adds noise to these geometric patterns until they resemble random static. In the reverse step, it learns to remove the noise step-by-step, guided by the protein’s amino-acid sequence, until coherent 3D-compatible geometries reappear, which can then be converted into full atomic models by standard structure-building software.
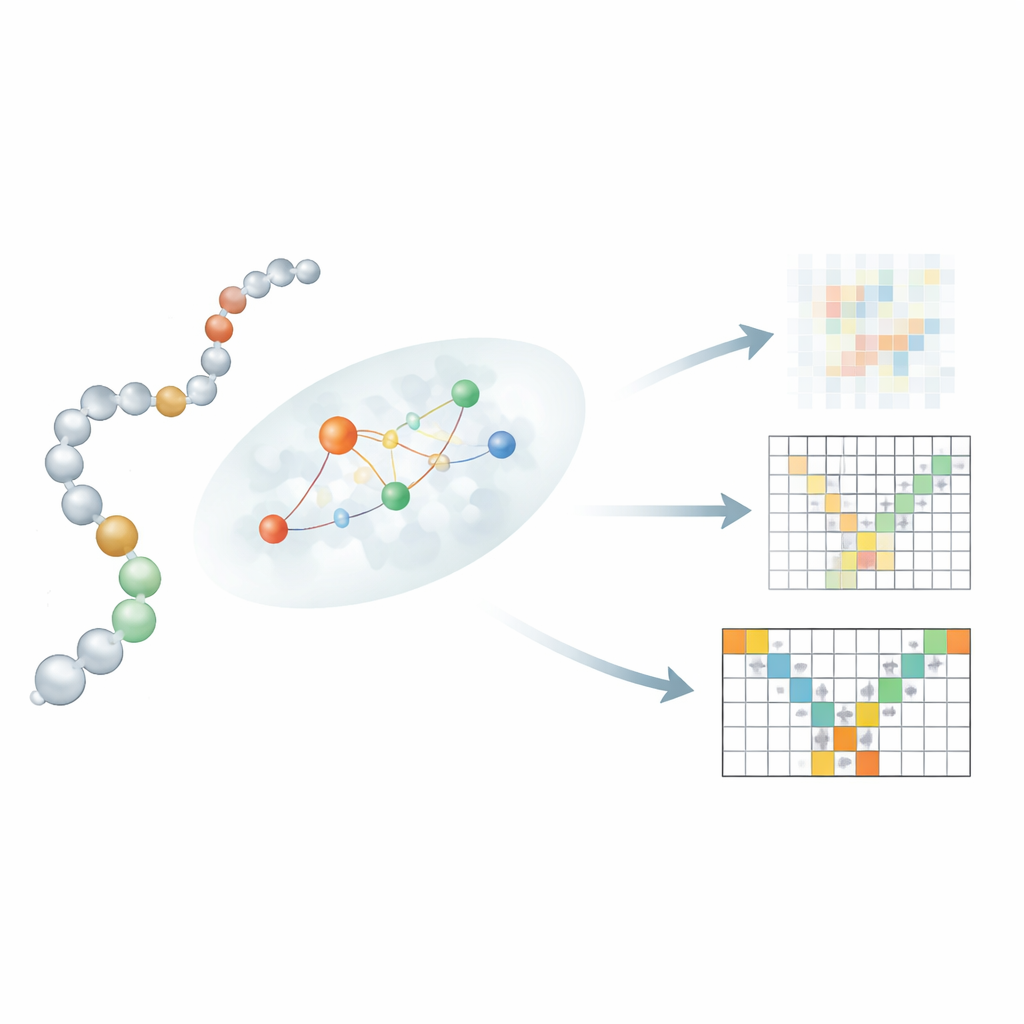
Letting sequence talk to structure locally
A key innovation lies in how Mac-Diff connects a linear sequence of residues to their 3D neighbors. Simply letting every residue attend to every other residue, as in text-to-image models, would blur important physical constraints. Instead, the authors introduce a "locality-aware" attention mechanism that focuses each residue on a small, probable neighborhood of interaction partners. To estimate these neighborhoods, Mac-Diff uses three ingredients: a protein language model called ESM-2 that encodes each residue’s biochemical context; a contact map hinting which pairs of residues are likely to be near each other; and a simple rule that favors residues close together along the chain. These signals are combined so that, during denoising, the model preferentially uses information from residues that are physically plausible partners, sharpening its ability to rebuild realistic, flexible structures.
Testing against long simulations and shape-changing proteins
The researchers tested Mac-Diff on two demanding fronts. First, they asked whether it could reproduce the broad distribution of shapes seen in long, carefully computed molecular dynamics simulations of fast-folding proteins and a classic benchmark protein known as BPTI. Across several measures that compare statistical properties of the generated ensembles to simulation data—such as distributions of distances inside the protein and overall compactness—Mac-Diff matched or surpassed competing AI methods, while also generating a wider variety of conformations. It captured most of the key “metastable” states identified in the simulations and reproduced residue-level flexibility patterns with high correlation, indicating that its ensembles reflect both global folds and local wiggles in a realistic way.
Revealing hidden functional states
Second, the team challenged Mac-Diff with proteins known to adopt very different shapes while doing their jobs, including the enzyme adenylate kinase, which toggles between open and closed forms during energy metabolism, and a curated set of 40 proteins with two experimentally determined conformations each. Mac-Diff generated only 100 candidate structures per protein—far fewer than typical simulation trajectories—yet still recovered most of the known states with good geometric agreement. In adenylate kinase, for instance, it produced both open and closed conformations with high similarity to crystal structures, whereas several popular methods tended to favor just one state. The model also ran about a thousand times faster than conventional simulations on comparable hardware, making systematic exploration of shape diversity far more practical.
What this means for biology and medicine
In everyday terms, Mac-Diff turns a protein’s sequence into a gallery of plausible poses rather than a single portrait, and it does so with an appreciation for which parts are likely to nudge or clasp each other in 3D. By accurately and efficiently sampling these ensembles, the method offers a way to probe how subtle shifts in shape underlie function, to spot rare yet important conformations, and to search for drug-binding pockets that appear only in transient states. While it does not yet capture the full time-ordered movies that simulations provide, Mac-Diff brings the dynamic landscape of proteins into reach for many more systems, promising new insights into structural biology, drug design, and protein engineering.
Citation: Wang, B., Wang, C., Chen, J. et al. Conditional diffusion with locality-aware modal alignment for generating diverse protein conformational ensembles. Nat Mach Intell 8, 415–434 (2026). https://doi.org/10.1038/s42256-026-01198-9
Keywords: protein dynamics, diffusion models, conformational ensembles, allosteric proteins, drug discovery