Clear Sky Science · en
Ectopic expression of cytosolic DHODH uncouples de novo pyrimidine biosynthesis from mitochondrial electron transport
Why breaking a hidden link in our cells matters
Every cell in your body must constantly copy and repair its DNA, a process that demands a steady supply of chemical “letters” called pyrimidines. In most animals, making these letters is tightly tied to how mitochondria—the cell’s power stations—burn fuel using oxygen. This coupling means that when mitochondrial respiration falters, DNA building blocks run short and cells struggle to grow. The study summarized here shows that borrowing a single gene from baker’s yeast can cleanly uncouple these two processes. That genetic tweak lets mammalian cells keep making DNA letters even when their mitochondria are impaired, opening a new way to probe, and perhaps one day treat, diseases driven by faulty energy metabolism.
A borrowed tool from yeast
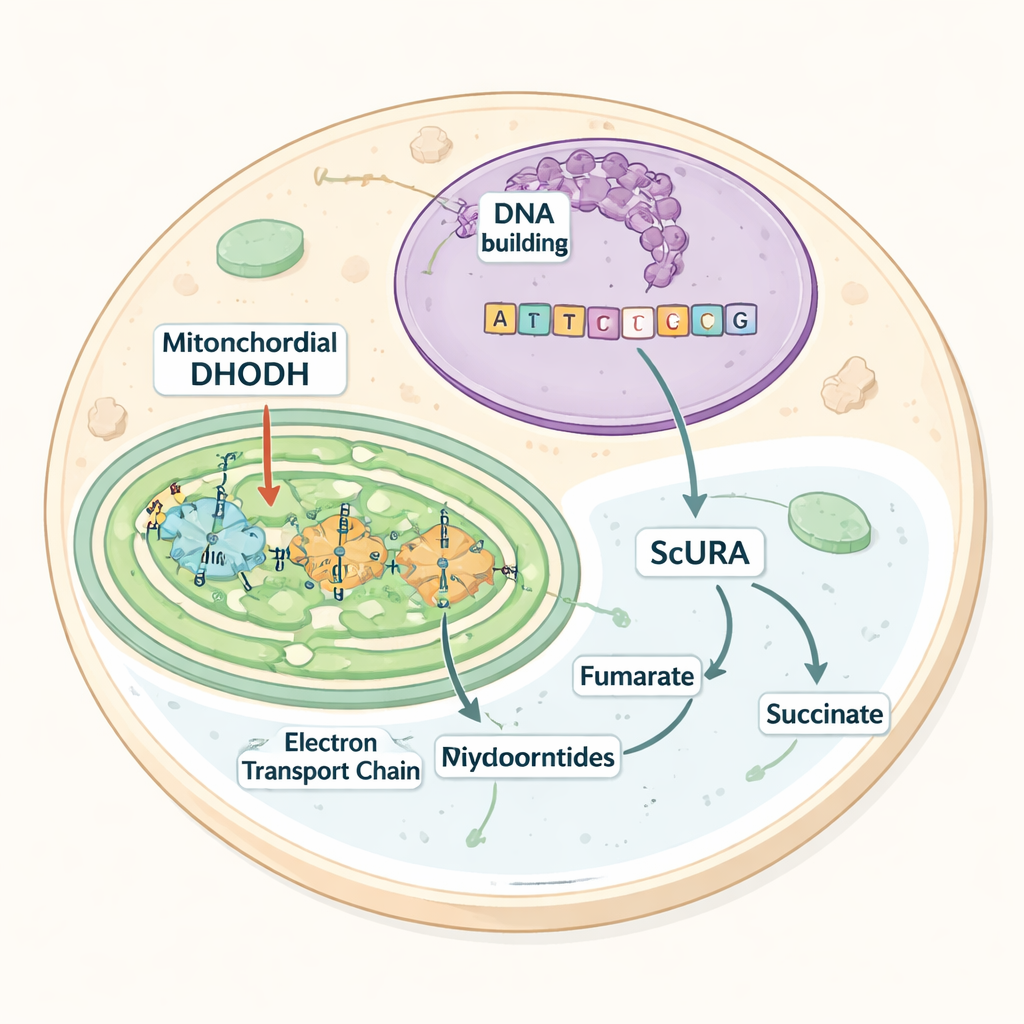
The work centers on an enzyme called dihydroorotate dehydrogenase (DHODH), which performs a key step in building pyrimidines. In mammals, DHODH sits on the inner membrane of mitochondria and hands electrons to a carrier molecule that feeds the respiratory chain. If this electron flow is blocked—for example by drug inhibitors or by genetic defects—DHODH stalls, pyrimidine production stops, and cells become dependent on dietary or supplied uridine, a ready-made building block. Many microbes that thrive without oxygen avoid this bottleneck by using alternative versions of DHODH that float in the cell fluid and use different electron acceptors. The authors asked whether they could install such an oxygen-independent route in mammalian cells.
Rewiring how DNA letters are made
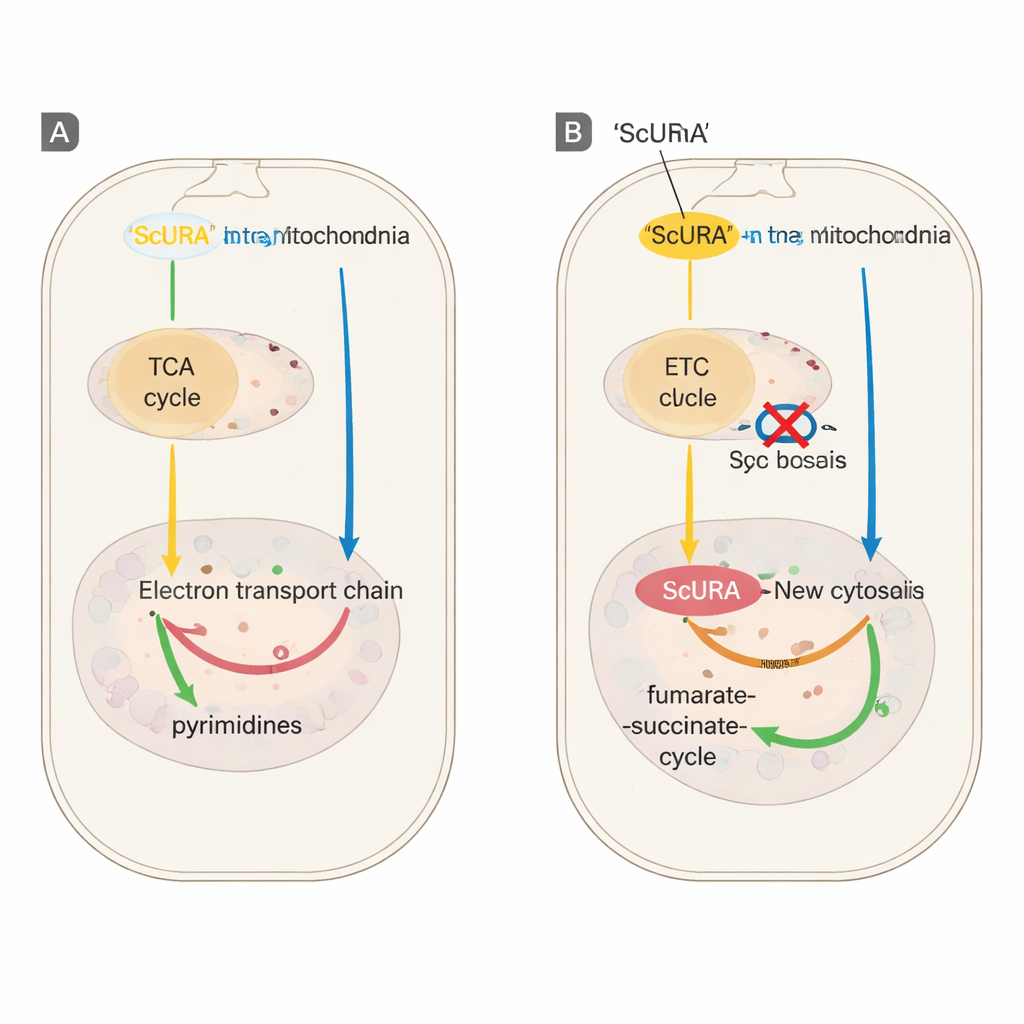
The researchers introduced the URA1 gene from the yeast Saccharomyces cerevisiae into human cells. URA1 encodes a cytosolic enzyme, dubbed ScURA, that performs the same chemical step as mitochondrial DHODH but hands electrons to a molecule called fumarate instead of to the mitochondrial carrier. Biochemical tests showed that ScURA formed active dimers in the cytosol and added a new, drug-resistant DHODH activity. Under normal conditions, adding ScURA did not disturb mitochondrial structure, respiration, or growth, indicating that it behaves as a backup pathway rather than an extra burden.
Cells that shrug off mitochondrial roadblocks
To see whether this backup could stand in for the native system, the team chemically blocked either DHODH itself or the mitochondrial electron transport chain, treatments that normally halt cell division unless uridine is supplied. Cells expressing ScURA continued to proliferate without any help, even under strong inhibition of respiratory complex III or after genetic deletion of the endogenous DHODH gene. Detailed tracing of nitrogen and carbon atoms from labeled glutamine showed that ScURA-expressing cells kept synthesizing pyrimidine nucleotides despite these blocks. Metabolite measurements revealed that, instead of accumulating toxic precursors, ScURA cells funneled electrons to fumarate, producing succinate and subtly reshaping the tricarboxylic acid (TCA) cycle to support a fumarate–succinate shuttle between cytosol and mitochondria.
Rescuing cells with damaged power plants
The authors then tested ScURA in cell models of mitochondrial disease. Cells lacking mitochondrial DNA, or carrying mutations that cripple respiratory complexes III or IV, normally rely on added uridine to grow. Once engineered to express ScURA, these diverse mutant cells were able to proliferate without uridine, although they still required pyruvate, reflecting a remaining need for some mitochondrial activity. At the gene-expression level, ScURA also prevented the shutdown of ribosomal protein genes that usually follows chronic electron transport inhibition, by keeping supplies of pyrimidine building blocks high enough to sustain RNA production.
What this means for health and disease
By cleanly separating pyrimidine synthesis from mitochondrial respiration, ScURA offers researchers a powerful new lever: they can now ask, in many settings, whether a given defect or drug effect truly stems from lost energy production or instead from a shortage of DNA and RNA precursors. In the long term, similar strategies might complement existing gene therapies for mitochondrial disorders, or help explain why some tumors depend so heavily on restoring their mitochondrial function. Although translating a yeast enzyme into human treatment will require great care, this study demonstrates that a single, well-chosen gene can rewrite a fundamental metabolic connection that evolution has long kept intertwined.
Citation: Curtabbi, A., Jaroszewicz, S.N., Sanz-Cortés, R. et al. Ectopic expression of cytosolic DHODH uncouples de novo pyrimidine biosynthesis from mitochondrial electron transport. Nat Metab 8, 454–466 (2026). https://doi.org/10.1038/s42255-026-01454-7
Keywords: pyrimidine metabolism, mitochondrial function, electron transport chain, metabolic rewiring, mitochondrial disease