Clear Sky Science · en
Machine learning-guided design of energy-related catalysts from nanoparticles to single-atom sites
Smarter Recipes for Cleaner Energy
Designing better catalysts—the tiny materials that speed up chemical reactions—is central to cleaner fuels, cheaper batteries, and greener industry. But finding the right recipe has long been a slow, trial-and-error process. This article explains how machine learning, the technology behind modern AI, is transforming that search. By teaching computers to spot patterns in vast troves of data, scientists can now home in on promising catalyst designs far faster, especially for cutting‑edge materials built from nanoparticles and even single metal atoms.
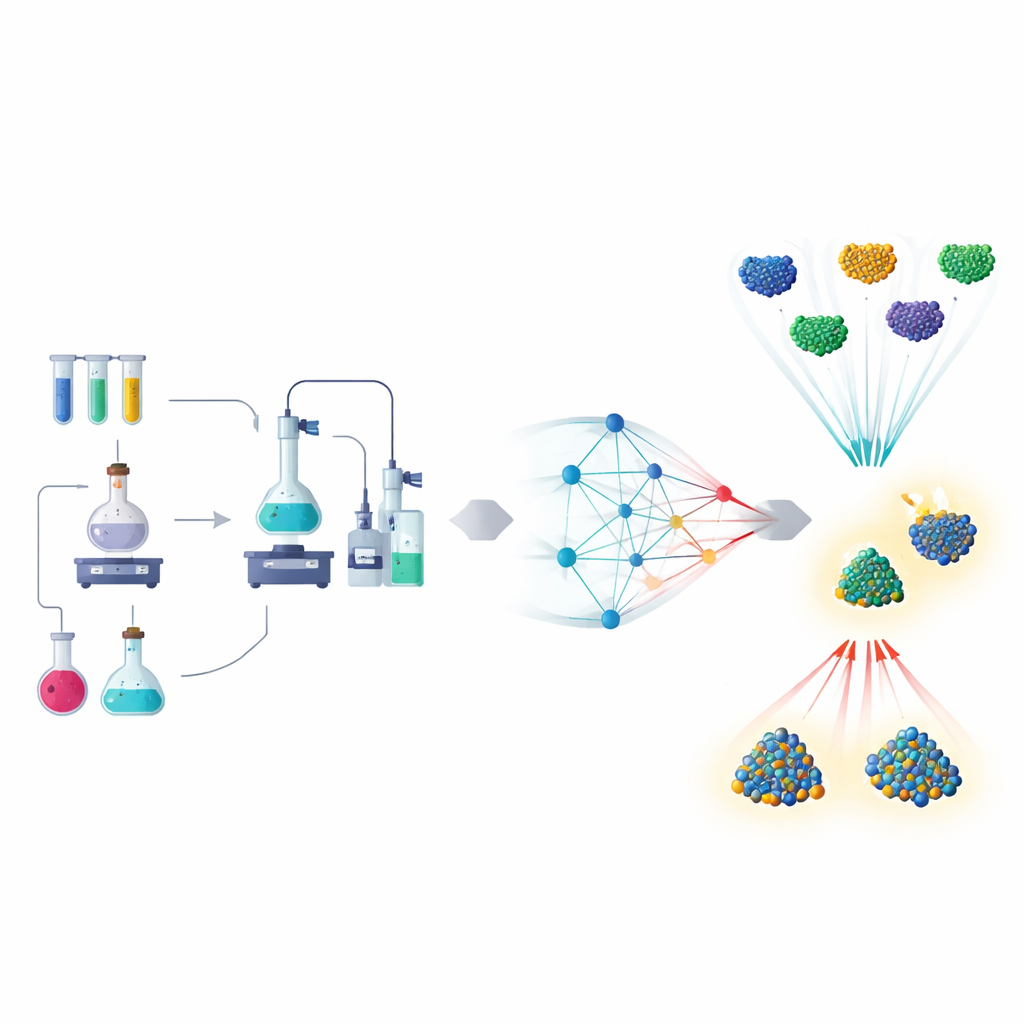
From Trial and Error to Data-Driven Discovery
Traditional catalyst research resembles cooking without a clear recipe: adjust a metal, change a support, tweak the temperature, then test and repeat. The paper describes how this approach is being reshaped by machine learning models that learn from both experiments and quantum‑level simulations. These models can predict how a catalyst will behave—how strongly it will bind key molecules, how fast reactions will run, or how long a material will last—without running every test in the lab. As a result, scientists can screen thousands of possibilities on a computer and reserve precious experimental time for only the most promising candidates.
Nanoparticles as a Testing Ground
Much of the early progress has come from nanoparticle catalysts, where tiny clusters of metal atoms carry out reactions such as splitting water or converting carbon dioxide. Here, machine learning uses simple inputs like particle size, surface structure, and composition to forecast performance. By digesting data gathered from years of experiments and simulations, these models can suggest which alloy combinations to try next, or which reaction conditions to explore. Automated robots, guided by these predictions, now run hundreds of experiments with little human intervention, dramatically speeding up the discovery of better materials for energy and environmental technologies.
Why Single Atoms Are So Special
The review then zooms in on single‑atom catalysts, where individual metal atoms are anchored on a solid support. These offer a tantalizing promise: every metal atom can be active, minimizing the use of costly elements like platinum or iridium. But because each atom sits in a unique local environment, their behavior is extremely sensitive to how they are bonded to nearby atoms. The authors show how machine learning helps decode this complexity. By feeding models simple numerical descriptors—such as how many electrons a metal has, how strongly it tends to attract other atoms, or how it is coordinated to its neighbors—researchers can map out how structure controls activity, selectivity, and stability for key reactions like oxygen evolution, fuel‑cell processes, nitrogen fixation, and carbon dioxide reduction.
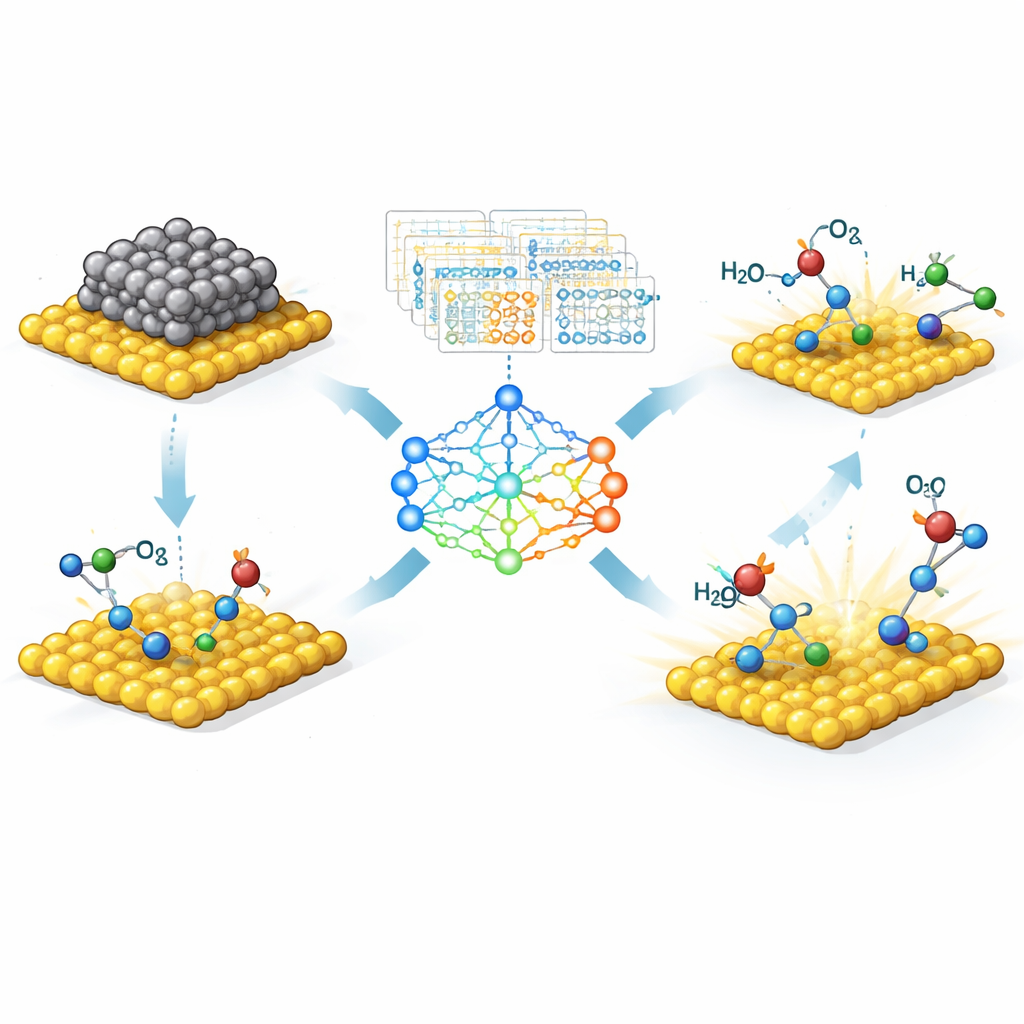
Finding Hidden Rules Behind Powerful Catalysts
A central theme of the article is the hunt for compact “descriptors,” simple combinations of basic properties that reliably forecast how a catalyst will perform. Machine learning helps sift through huge sets of possibilities to identify a small handful that matter most, turning messy data into clear design rules. For example, the number of electrons in particular orbitals of the metal atom, or how charge is shared between the metal and its support, can often predict how strongly crucial reaction intermediates will bind. In some cases, these rules can be captured in short equations that scientists can apply directly to screen thousands of potential single‑atom or dual‑atom catalysts on a computer before making them in the lab.
Making Sure Catalysts Last
Good catalysts must not only be active; they must also be durable. The review describes how machine learning models can estimate whether single atoms will stay put on their supports or clump together into less effective particles. By relating the strength of the metal–support bond and the metal’s own cohesion to how quickly atoms are likely to diffuse and aggregate, the authors show that stability can be predicted from a few basic numbers. This allows researchers to filter out fragile designs early and focus on materials that can survive harsh industrial conditions, such as high temperatures or corrosive solutions.
Where AI-Guided Catalysts Are Headed Next
Looking forward, the paper argues that the full power of machine learning in catalyst design will come from three advances: better shared databases, smarter and more transparent models, and closer ties to real‑world conditions. Large, standardized collections of experimental and computational data will allow algorithms to learn more general rules rather than case‑by‑case tricks. New “white‑box” models that blend physics with data science could provide both accuracy and insight, avoiding black‑box predictions that are hard to trust. Finally, by feeding models with data from pilot plants and operating devices, researchers hope to optimize catalysts not just for ideal laboratory tests, but for long‑term, cost‑effective performance in working energy technologies.
Citation: Hu, Z., Wang, Z., Peng, Y. et al. Machine learning-guided design of energy-related catalysts from nanoparticles to single-atom sites. Commun Chem 9, 128 (2026). https://doi.org/10.1038/s42004-026-01967-y
Keywords: machine learning catalysts, single atom catalysts, nanoparticle catalysis, energy conversion materials, data-driven materials design