Clear Sky Science · en
Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature
Why this matters for everyday chemistry
Computer simulations are the workhorses of modern chemistry and materials science. They let scientists watch molecules twist, vibrate, and collide in silico instead of in expensive, time‑consuming experiments. But when these simulations rely on machine learning, they can suddenly "blow up," producing impossible molecular shapes—especially at higher temperatures. This study introduces a new kind of physics‑informed machine‑learning model that can run such simulations for extremely long times, at temperatures up to 1000 kelvin, without falling apart.
From clever shortcuts to fragile simulations
Traditional quantum chemistry calculates molecular energies with high accuracy but is painfully slow. Simpler force fields are fast but often approximate. Machine‑learned potentials aim to combine the best of both worlds: they learn a shortcut from molecular geometry to energy and forces, then use that shortcut to drive molecular dynamics. On paper, many such models look excellent, boasting tiny average errors on standard test sets. In practice, those numbers can be deceptive. When molecules explore new shapes during a simulation—especially at higher temperatures—many models are pushed outside the range of structures they were trained on. Instead of gently steering molecules back to realistic shapes, they may predict forces that stretch or crush bonds until the entire system becomes unphysical and the simulation crashes.
Building models on quantum‑based building blocks
The authors tackle this fragility by changing what the model learns and how it is guided by prior physics. They use a framework called FFLUX, which builds on the Interacting Quantum Atoms (IQA) approach. In IQA, a molecule is divided into "topological atoms" whose individual energies are determined directly from quantum mechanics. These atomic energies are physically meaningful and add up to the total energy of the molecule. Instead of learning arbitrary site energies, the new Gaussian process models learn these quantum‑rooted atomic energies, providing a deep physical anchor for every prediction. Four flexible organic molecules—peptide‑capped glycine and serine, malondialdehyde, and aspirin—serve as challenging test beds because of their many internal motions and known difficulty for existing machine‑learning force fields.
Teaching the model to expect trouble
A key innovation lies in how the Gaussian process is set up before it ever sees data: its "mean function," which encodes what the model assumes in poorly known regions. Most previous work simply sets this mean to zero, effectively pretending that the model has no prior expectations. The authors instead deliberately shift this mean toward higher atomic energy states, while keeping it physically sensible. This design choice means that when the model is forced to extrapolate—say, when bonds are temporarily overstretched—it naturally prefers predictions that penalize extreme distortions. In extensive tests, versions of the model differing only in this prior behaved very differently. Models with conventional or low‑energy means often survived less than a picosecond before a molecule exploded or imploded. In contrast, the best high‑energy mean (dubbed MF5) yielded simulations that remained stable for the full nanosecond test window at temperatures from 300 to 1000 kelvin for all four molecules.
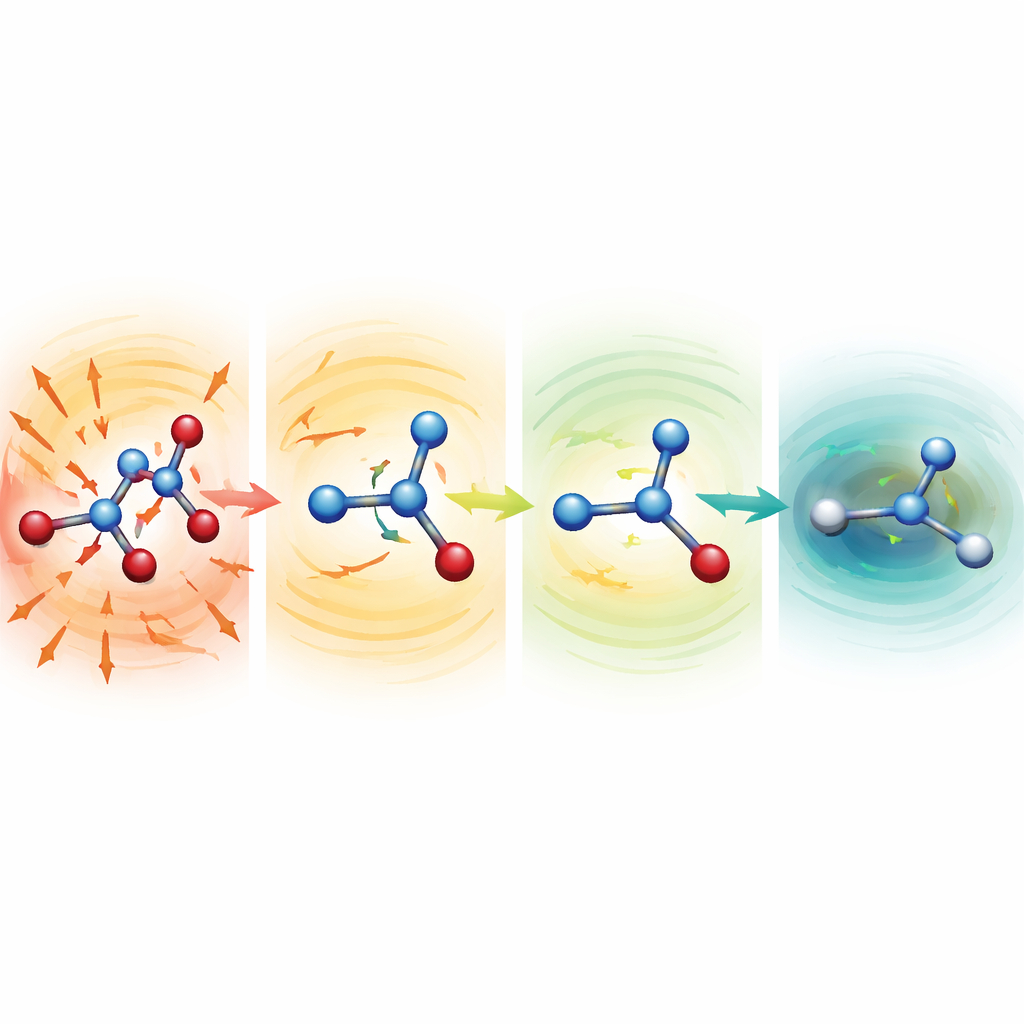
Watching distorted molecules heal themselves
To probe why the robust models work so well, the researchers started simulations from deliberately mangled structures with severely stretched or compressed bonds. For serine, aspirin, and malondialdehyde, these starting points lay hundreds to more than a thousand kilocalories per mole above a normal structure—configurations that would normally be disastrous. With weaker mean functions, the molecules quickly flew apart. With the MF5 setup, however, the predicted forces immediately pointed in directions that shortened elongated bonds and lengthened squashed ones. Within tens to hundreds of time steps, the molecules relaxed into realistic shapes and then continued to evolve stably. The team also showed that, without ever being trained on forces, the same models can guide geometry optimizations of alanine dipeptide, reproducing known low‑energy conformations and relative energies within a few tenths of a kilocalorie per mole, but at around 200‑fold lower cost than full quantum calculations.
Long, hot simulations on everyday hardware
Robustness is not just about surviving one difficult start; it is about holding up over millions or billions of time steps. The authors pushed their best models further by running 50 independent simulations at 500 kelvin, each for 10 nanoseconds, across their four test molecules. None of these runs crashed, yielding a combined simulation time of half a microsecond—unusual for cutting‑edge machine‑learned force fields. Even more striking, the simulations ran efficiently on standard CPUs, step‑for‑step rivaling or beating some prominent neural‑network potentials that require powerful GPUs. Throughout, the molecules explored rich sets of shapes and metastable states, showing that robustness was not achieved by artificially freezing motion or enforcing rigid structures.
What this means for future molecular modeling
For non‑experts, the key message is that not all low‑error machine‑learning models are trustworthy when pushed hard. By grounding their models in quantum‑derived atomic energies and carefully tuning the model’s built‑in expectations toward high‑energy states, the authors created a family of potentials that naturally produce "restoring forces"—the molecular equivalent of a safety harness—that keep simulations physical even at high temperatures and from distorted starting points. This approach promises more reliable and longer simulations of complex molecules, and it points toward future extensions where similar physics‑informed models handle condensed phases and subtle interactions like dispersion, all while remaining computationally practical.
Citation: Isamura, B.K., Aten, O., Nosratjoo, M. et al. Unprecedented robustness of physics-informed atomic energy models at and beyond room temperature. Commun Chem 9, 138 (2026). https://doi.org/10.1038/s42004-026-01965-0
Keywords: machine learning force fields, molecular dynamics robustness, Gaussian process potentials, physics-informed modeling, quantum atomic energies