Clear Sky Science · en
Fine-tuning AlphaFold with limited cryo-EM observations
Why protein shapes are so hard to see
Proteins are tiny molecular machines that drive nearly every process in our bodies, from energy production to nerve signals. To understand how they work—and how drugs might control them—scientists need to know their precise three-dimensional shapes. Two powerful tools have emerged for this job: cryo–electron microscopy (cryo‑EM), which takes many blurry snapshots of frozen proteins, and AlphaFold, an artificial‑intelligence system that predicts protein structures from their sequences. But in many real experiments the cryo‑EM data are incomplete, and AlphaFold’s predictions do not always match reality. This paper introduces CoCoFold, a method that teaches AlphaFold to listen directly to difficult cryo‑EM data and improve its predictions accordingly.
When the camera sees too little
Cryo‑EM works by flash‑freezing proteins and imaging enormous numbers of individual particles from many angles, then combining those images into a 3D map. In practice, however, researchers often do not have enough good images to work with. Sometimes the protein appears only briefly in a high‑energy state, so very few particles are captured. In other cases, proteins prefer certain orientations on the ice surface, so many viewing angles are missing. Both problems lead to fuzzy, incomplete maps that are hard to translate into reliable atomic models. Existing software can fit AlphaFold’s predicted structures into such maps, but its success depends heavily on having sharp, high‑resolution data to begin with.
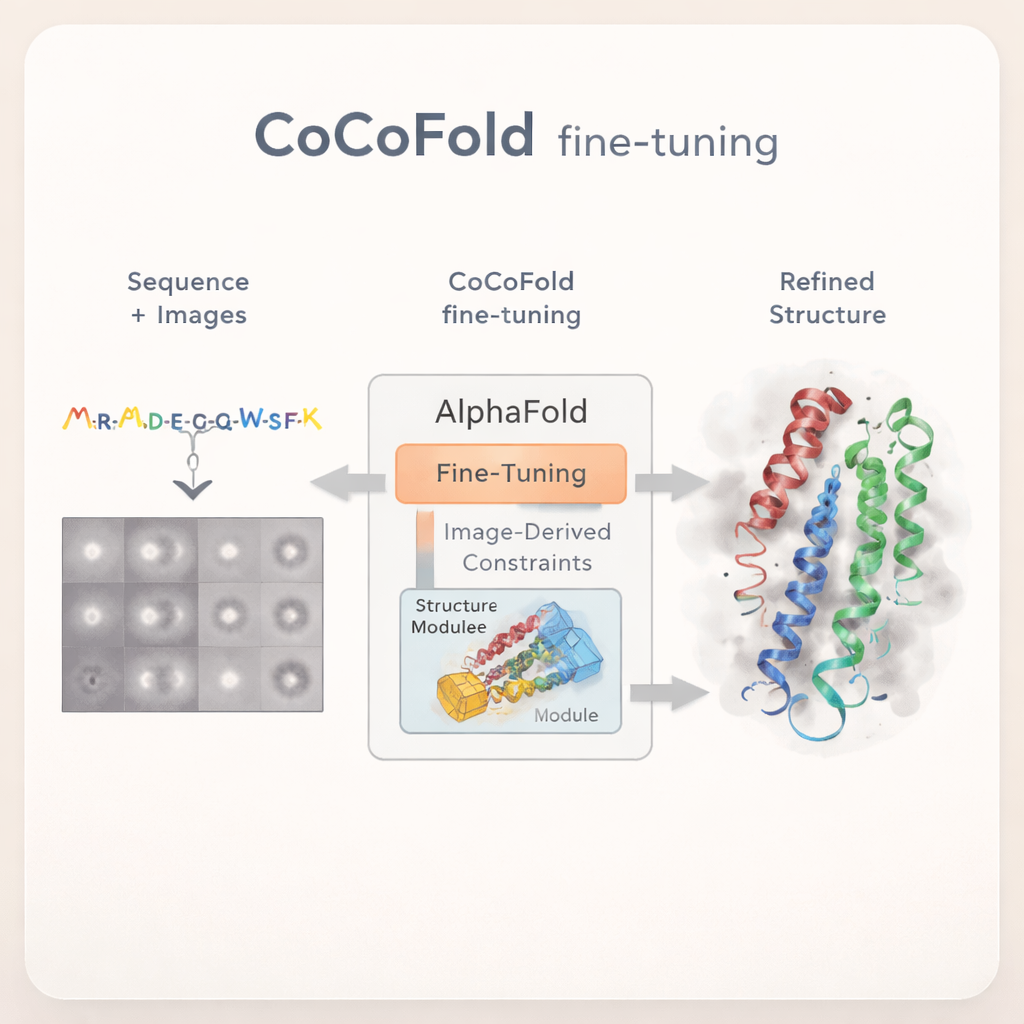
Teaching AlphaFold to learn from raw images
CoCoFold takes a different approach: instead of relying on a fully reconstructed 3D cryo‑EM map, it directly uses the raw 2D particle images to fine‑tune AlphaFold. The method starts from an AlphaFold‑Multimer prediction and keeps most of the original network frozen, preserving its broad knowledge of protein folding. Only the final structure‑building part is allowed to change. A lightweight "adapter" is added to feed information derived from the cryo‑EM images into this structure module, gently nudging the model toward shapes that are compatible with the experimental data while avoiding wild deviations from known protein physics.
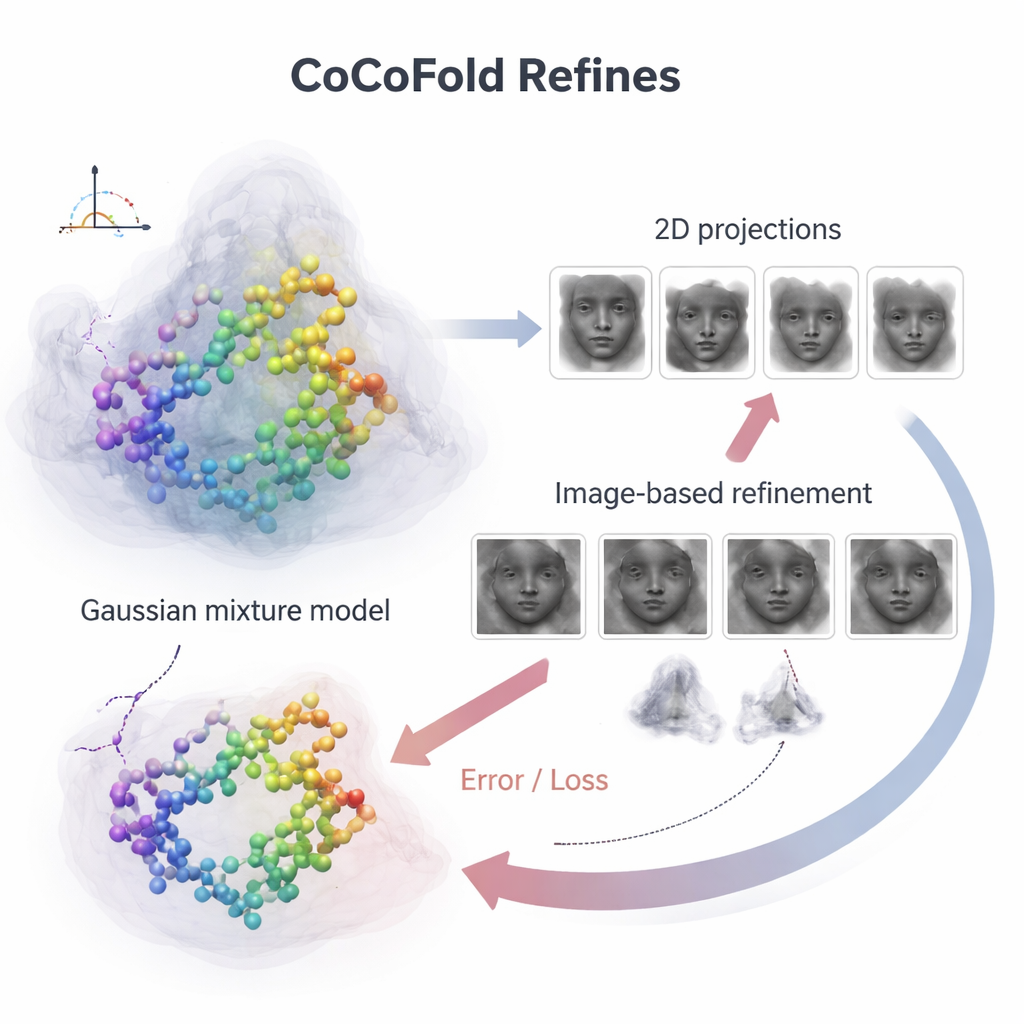
Turning images into structural feedback
To connect individual protein atoms to the noisy microscope images, CoCoFold builds a smooth, flexible picture of the predicted structure using overlapping three‑dimensional blobs, known as a Gaussian mixture. From this representation it simulates how the protein would look in the microscope at the same viewing directions and imaging conditions as the real experiment. These simulated snapshots are then compared to the actual cryo‑EM particles, ring by ring in the frequency domain, to see how well they match. Any mismatch becomes a feedback signal that flows back through the network, slightly adjusting both the protein model and the density representation. After training, the atomic model is further cleaned up with a physics‑based refinement step to remove local geometric clashes.
Staying accurate when data are scarce or biased
The authors tested CoCoFold on several experimental and simulated datasets designed to mimic the two main problems in cryo‑EM: too few particles and large gaps in viewing angles. Under these tough conditions, standard tools—including other deep‑learning methods that depend on reconstructed maps—tended to miss regions of the protein, misplace helices, or lose fine details as the maps became blurrier. CoCoFold, by contrast, consistently produced models that matched known reference structures more closely and more completely. Its errors remained small even when the number of particles was drastically reduced or when large cones of viewing directions were missing, suggesting that directly learning from the raw images preserves crucial information that map‑based approaches discard.
What this means for future structural biology
For non‑specialists, the key message is that CoCoFold acts like a translator between powerful AI predictions and imperfect experimental data. Instead of trusting either AlphaFold or cryo‑EM alone, it lets the two inform each other, especially in the difficult regimes where experiments provide only a partial view. In straightforward cases with abundant, high‑quality data, existing map‑driven tools still work extremely well. But when particles are rare or orientations are missing—common situations when chasing fleeting or fragile protein states—CoCoFold offers a way to recover reliable atomic models from information that would otherwise go to waste.
Citation: Liao, J., Zheng, D., Zhang, H. et al. Fine-tuning AlphaFold with limited cryo-EM observations. Commun Chem 9, 95 (2026). https://doi.org/10.1038/s42004-026-01899-7
Keywords: cryo-EM, AlphaFold, protein structure, deep learning, structural biology