Clear Sky Science · en
NRF2 activators and the inhibitor of nuclear export, selinexor, restrict coronaviruses by targeting a network involving ACE2, TMPRSS2, and XPO1 through an NRF2-independent mechanism
Drugs That Help Cells Say “No” to Coronaviruses
Most coronavirus treatments today go after the virus itself. But viruses are masters of change, and new variants can quickly blunt those drugs. This study explores a different strategy: helping our own cells become less welcoming to both dangerous coronaviruses like SARS-CoV-2 and milder seasonal strains. By tweaking how cells handle a few key gatekeeper proteins, the researchers show that existing small molecules can sharply reduce infection in laboratory models, and they do so in a way that doesn’t rely on the usual antiviral pathway they were famous for.
A New Angle on Coronavirus Defense
The team focused on compounds known for activating a cellular protection program controlled by a protein called NRF2. These compounds—4-octyl itaconate (4OI), bardoxolone (BARD), and sulforaphane (SFN)—were tested alongside selinexor (SEL), a drug that blocks a transport protein called XPO1, which helps move other proteins out of the cell nucleus. In lung and kidney cell models, all four drugs cut SARS-CoV-2 levels without harming the cells. They also worked against several variants of concern. Surprisingly, when NRF2 was genetically removed, the viruses actually replicated better, proving that NRF2 is naturally protective—but the drugs still worked well, showing that their antiviral power comes from a different route. 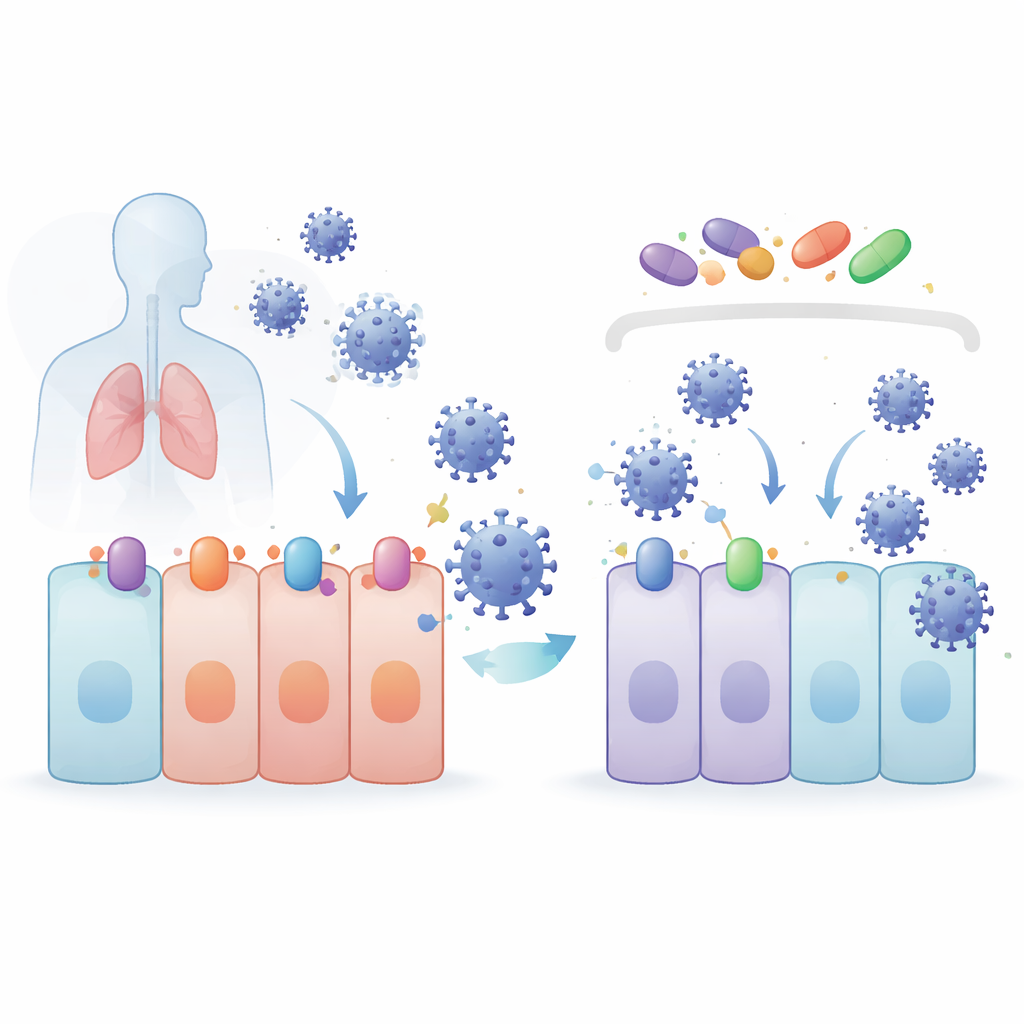
Shutting the Viral Front Door
Coronaviruses start infection by docking onto receptor proteins on the cell surface. For SARS-CoV-2 and its close relatives, the main door is ACE2, with help from a cutter enzyme called TMPRSS2. The export protein XPO1 also seems to aid infection. The researchers found that 4OI, BARD, SFN, and SEL all lowered the amounts of ACE2, TMPRSS2, and XPO1 in human lung cells. 4OI and SEL were especially powerful when present before infection, and they blocked the entry of virus-like particles coated with coronavirus spike proteins. In other words, these drugs don’t just slow the virus after it gets in—they help remove the locks and handles the virus needs to open the door in the first place.
How Cells Dismantle Key Viral Aids
Digging deeper, the team showed that 4OI speeds up the breakdown of ACE2 protein. Under normal conditions, ACE2 lasts for many hours; with 4OI, it disappeared from the cell surface in a fraction of that time. This destruction required two cellular taggers, NEDD4L and MDM2, which attach molecular “dispose me” flags to proteins. Blocking these taggers weakened 4OI’s ability to eliminate ACE2. Surprisingly, the usual protein-shredding machinery, the proteasome, was not the main route. Instead, ACE2 was funneled into the cell’s recycling and waste system, the lysosome. The drugs also lowered the gene activity for ACE2 and XPO1, in part by reducing activation of STAT3, a protein that normally boosts ACE2 gene reading. 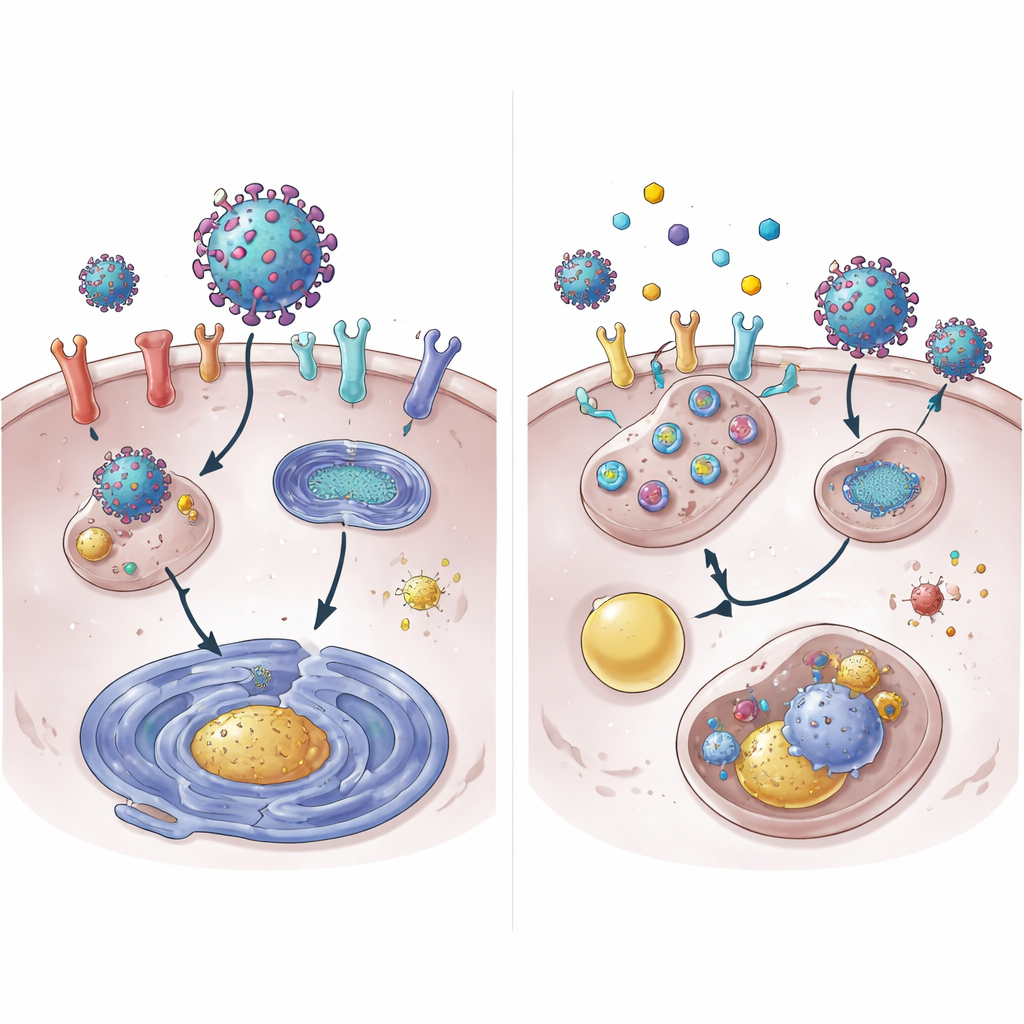
Seasonal Coronaviruses and the XPO1 Connection
The scientists next turned to hCoV-229E, a seasonal coronavirus that usually causes mild colds but can be dangerous in people with weak immune systems. Using engineered viruses that glow when they replicate, they showed that all four compounds strongly suppressed 229E in lung and blood vessel cells, even when NRF2 was absent. Unlike SARS-CoV-2, this virus uses a different receptor, ANPEP, which the drugs did not change. Instead, their impact tracked closely with how strongly they reduced XPO1. Knocking down XPO1 itself sharply lowered 229E replication, and selinexor’s effect almost vanished in these XPO1-poor cells. This ranking—SEL most dependent on XPO1, BARD least—suggests each compound has a slightly different mix of targets, with XPO1 as a central hub for many of their antiviral actions.
What This Could Mean for Future Treatments
For non-specialists, the key message is that it is possible to fight coronaviruses not only by attacking the virus, but by gently rewiring our own cells so they are harder to infect. In lab-grown human cells, the studied compounds stripped away crucial docking sites and helper pathways that SARS-CoV-2 and a common cold coronavirus rely on, and they did this largely without the very NRF2 pathway that first made them interesting. While these findings are still preclinical and do not yet translate directly into medicines, they highlight a promising path: drugs that simultaneously lower viral entry, dampen harmful inflammation, and protect tissues by targeting a shared network of host proteins such as ACE2, TMPRSS2, and XPO1.
Citation: Waqas, F.H., Silva da Costa, L., Zapatero-Belinchón, F.J. et al. NRF2 activators and the inhibitor of nuclear export, selinexor, restrict coronaviruses by targeting a network involving ACE2, TMPRSS2, and XPO1 through an NRF2-independent mechanism. Commun Biol 9, 384 (2026). https://doi.org/10.1038/s42003-026-09724-6
Keywords: host-directed antivirals, coronavirus entry, ACE2 and TMPRSS2, NRF2 activators, XPO1 inhibition