Clear Sky Science · en
Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy
When Pain Nerves Run Out of Power
Some people are born almost unable to feel pain. At first glance this might sound like a blessing, but it quickly becomes a curse: without pain as a warning signal, they accumulate burns, fractures, infections and even blindness. This study investigates one rare form of such pain-loss disorders and uncovers a surprising culprit: tiny power plants inside nerve cells whose energy production goes badly awry.
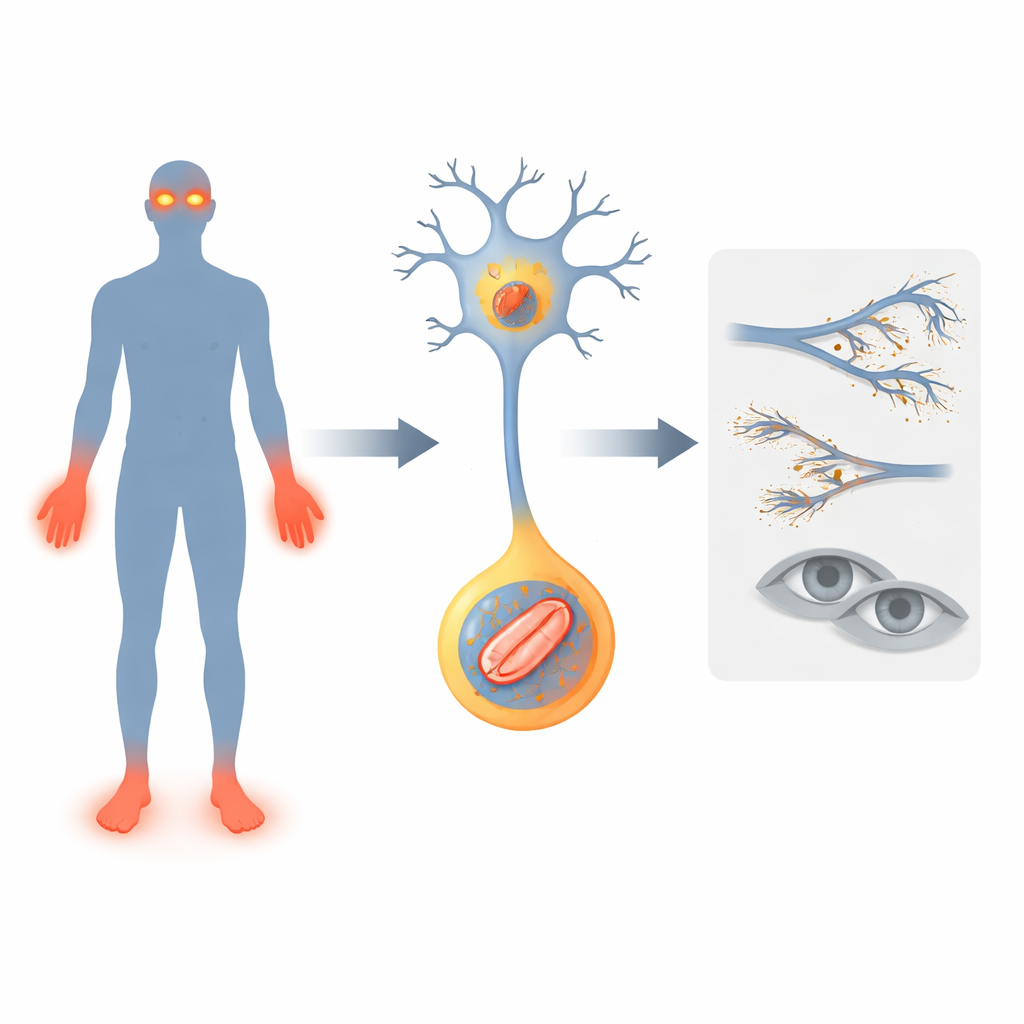
A Gene That Silences the Alarm Bells
The researchers focus on a gene called FLVCR1, already linked to rare nerve conditions where people lose pain sensation, develop unsteady walking and sometimes progressive vision loss. They describe two new patients with changes in FLVCR1. Both children showed early problems: delayed motor milestones, frequent falls, deep infections and mutilation of fingers and toes because injuries went unnoticed. One also developed a degenerative eye disease called retinitis pigmentosa, leading to night blindness. These cases broaden the picture of how FLVCR1 defects can manifest in humans and strengthen the idea that this gene is vital for keeping pain-sensing nerves and light-sensing cells in the retina alive.
Modeling the Disease in Tiny Fish
To explore how FLVCR1 affects developing sensory nerves, the team turned to zebrafish, whose transparent embryos allow direct viewing of nerve cells. They reduced levels of the fish version of the gene, flvcr1a, using genetic tools. Fish with lowered flvcr1a had fewer dorsal root ganglia, clusters of neurons that detect touch and pain along the spine. Behaviorally, these fish moved less on their own and swam only short distances when their tail was gently touched, suggesting a dulled sensory response. Because earlier mouse models died too early to analyze their sensory nerves, these zebrafish provide the first living system in which FLVCR1-related nerve defects and behavior can be followed in detail.
Three Troubled Pathways Converge on the Cell Powerhouses
FLVCR1 sits in cell membranes and manages several key substances. Previous work suggested roles in handling choline (a building block of membrane lipids), heme (the iron-containing pigment that powers many enzymes) and calcium flow between cell compartments. The scientists collected skin cells (fibroblasts) from four patients carrying different FLVCR1 mutations and compared them with cells from healthy people and symptom-free carriers. They found that patient cells had lower choline levels and more fluid cell membranes, changes that could disturb the delicate lipid environment required by mitochondria, the energy-generating organelles. They also discovered that a crucial enzyme for making heme inside mitochondria, ALAS1, was less active, even though the total heme content looked almost normal. At the same time, the physical contact sites between the endoplasmic reticulum and mitochondria—where calcium normally flows into mitochondria—were shorter and less frequent, and calcium entry into mitochondria was reduced. Three problems—choline shortage, sluggish heme production and weakened calcium transfer—all pointed toward impaired mitochondrial performance.
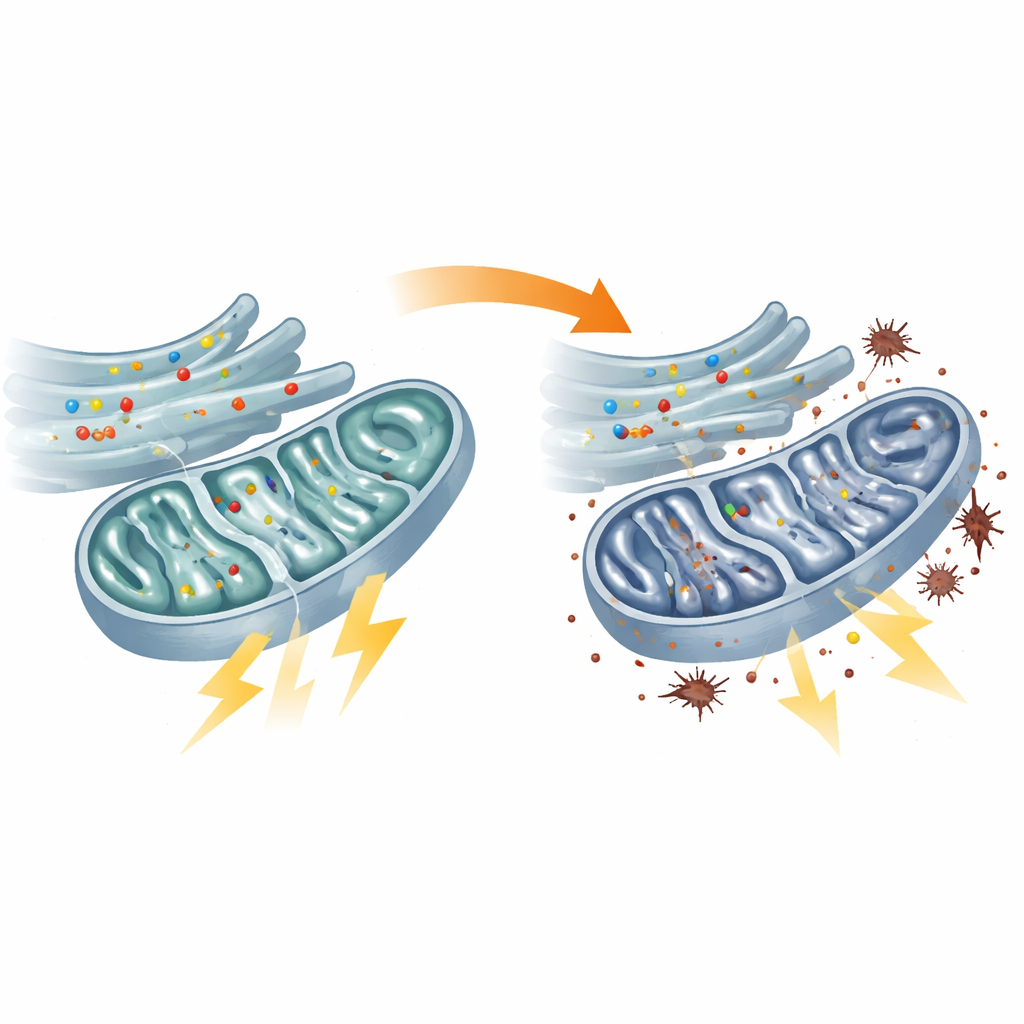
Starved Mitochondria and Overworked Backup Systems
Direct tests of energy metabolism confirmed that the mitochondria in patient fibroblasts were underperforming. The central fuel-processing hub known as the TCA cycle ran more slowly, several of its key enzymes were less active and the chain of reactions that normally converts fuel into ATP, the cell’s energy currency, was blunted. As a result, ATP levels inside mitochondria fell. The cells tried to compensate by ramping up glycolysis, a less efficient, sugar-burning pathway outside mitochondria. This shift in energy strategy came at a cost: electrons leaked from the stressed mitochondrial machinery and triggered higher levels of lipid peroxidation, a form of oxidative damage to cell membranes. Similar defects were seen in zebrafish with reduced flvcr1a, tying mitochondrial failure directly to the animal model of sensory neuropathy.
Hints of Future Treatments by Boosting Cell Energy
Encouragingly, some of these defects could be eased in the lab. When the team artificially increased calcium entry into mitochondria by overproducing a channel protein called MCU in patient cells, energy production rebounded and signs of oxidative damage dropped. Supplying cells with a precursor of heme, 5-aminolevulinic acid (ALA), also improved TCA cycle activity, respiratory chain function and ATP levels, although prolonged ALA exposure was harmful in previous studies. Extra choline normalized membrane fluidity and helped reduce lipid damage, but gave only modest short-term gains in energy output. These rescue experiments suggest that no single pathway is solely responsible; instead, a network of disrupted choline, heme and calcium handling pushes mitochondria into chronic underperformance.
Why These Findings Matter for Patients
By tracing the consequences of FLVCR1 mutations from molecules to cells to whole organisms, this work proposes that energy failure in mitochondria is a driving force behind this form of pain-loss neuropathy and its associated vision problems. Sensory nerves and photoreceptors are unusually energy-hungry because they maintain long axons or continuously renew light-sensitive structures, making them especially vulnerable when mitochondrial output falters. The study’s zebrafish model and patient-derived cells now offer practical testing grounds for therapies that strengthen mitochondrial metabolism. While treatments such as choline supplementation, controlled heme boosting or drugs that enhance mitochondrial calcium uptake will require careful evaluation in animal models and clinical trials, the central message is clear: restoring the power supply of fragile neurons may one day help protect people born without nature’s most important warning signal—pain.
Citation: Bertino, F., Zanin Venturini, D.I., Grasso, E. et al. Mitochondrial energetic failure underlies FLVCR1-related sensory neuropathy. Commun Biol 9, 429 (2026). https://doi.org/10.1038/s42003-026-09691-y
Keywords: sensory neuropathy, mitochondrial dysfunction, FLVCR1, pain insensitivity, nerve energy metabolism