Clear Sky Science · en
A practical guide to targeted single-cell RNA sequencing technologies
Why looking at single cells matters
Every cell in your body carries the same DNA, yet different cells behave in very different ways. They do this by turning specific genes on or off and by editing RNA molecules in subtle ways. Modern single-cell RNA sequencing can read which RNAs are present in thousands of cells at once, but it currently misses most of the message. This review explains where today’s techniques lose information and how new “targeted” methods are being developed to zoom in on the most important parts of RNA molecules for research, diagnosis, and treatment design.
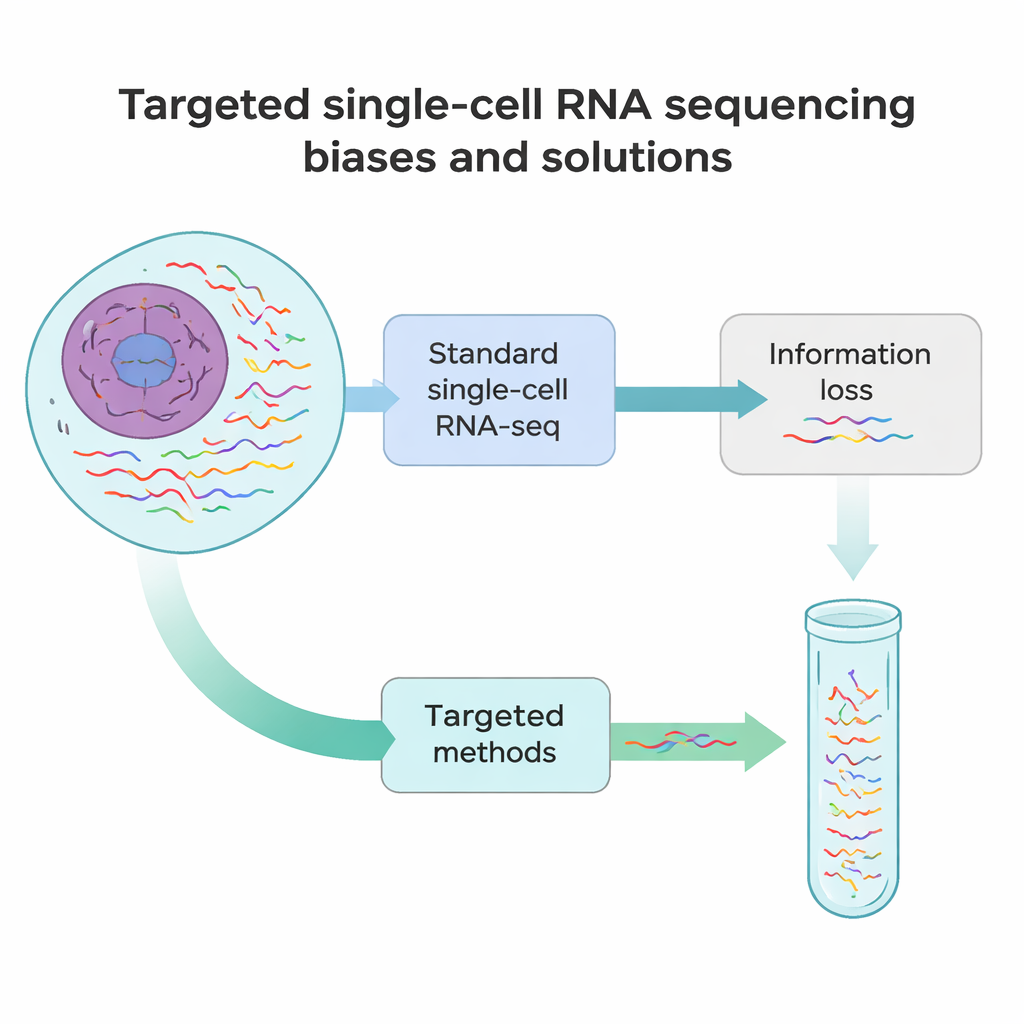
Where today’s methods fall short
Standard single-cell RNA sequencing works a bit like taking a quick snapshot of each message in a cell rather than a full-length movie. In most experiments, only about 10–40% of all RNAs in a cell are detected, and only their beginning or end is read. That means many rare but important RNAs—such as markers that define a cell’s identity, or gene versions carrying disease-causing mutations—are easily missed. On top of that, several technical steps, from breaking tissues into single cells to copying RNA into DNA and amplifying it, introduce systematic biases. Some RNAs are cut off early, some are overrepresented, and others vanish from the data entirely.
Why internal RNA details matter
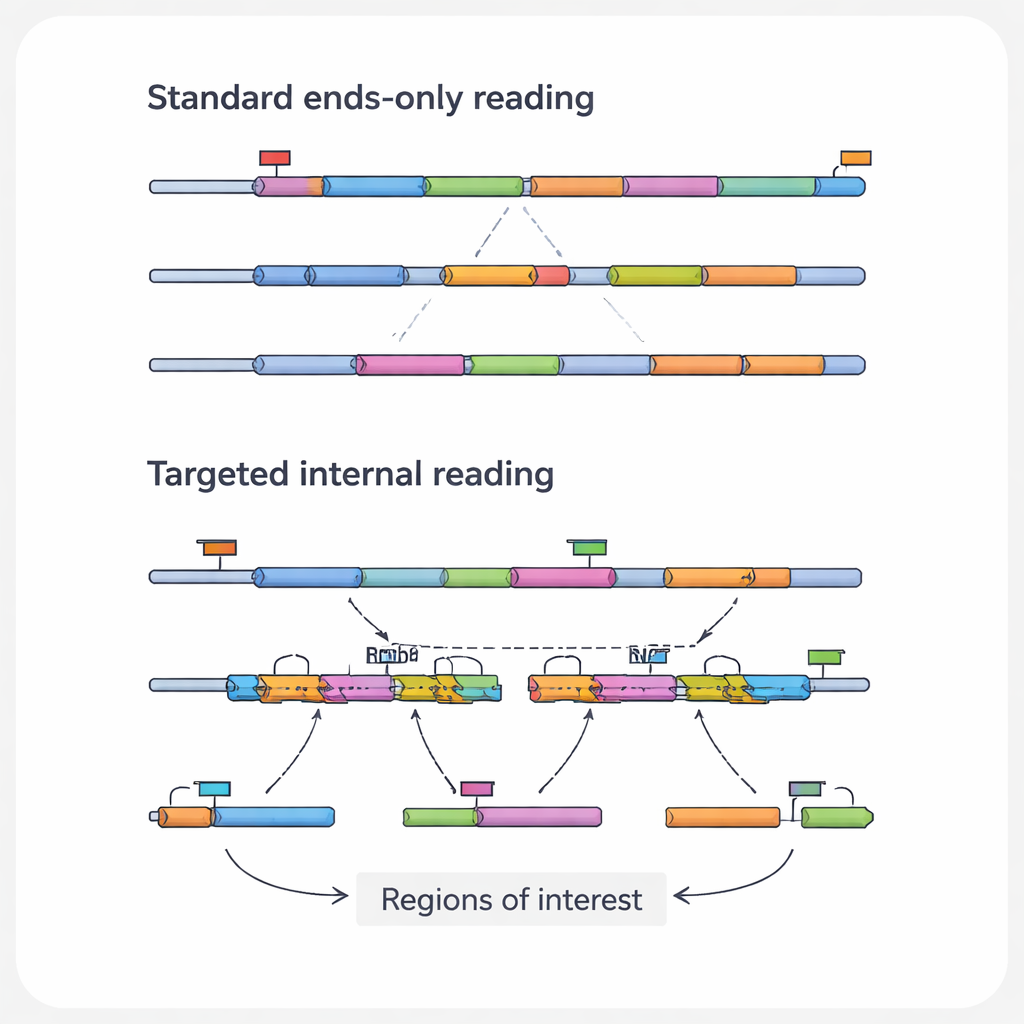
The most medically relevant information in an RNA molecule often lies in its internal regions, not at the ends that standard methods see. These inner sections can contain point mutations that drive cancer, fusion points where two genes have been abnormally joined, or splice junctions that create different protein variants from the same gene. They can also record the footprints of gene-editing tools such as CRISPR. The authors call these specific features “regions of interest,” and the RNAs that carry them “transcripts of interest.” Because common high-throughput platforms mainly read the tips of RNAs, they routinely overlook these crucial details, especially in long or low-abundance transcripts.
New ways to aim the spotlight
To overcome these blind spots, researchers have developed a family of targeted single-cell RNA sequencing approaches. Instead of trying to read every RNA equally, these methods deliberately enrich for selected transcripts or regions. Some strategies redesign the capture beads so they latch onto internal RNA sequences rather than just the tail, pulling chosen messages into the library from the very first step. Others add custom primers that start copying at an internal point, or extra PCR steps that specifically amplify a shortlist of genes out of an existing library. Yet another group uses DNA probes that hybridize to target RNAs or their copies and then fish them out, often with simple chemical tags. Each category trades off sensitivity, number of cells, number of targets, and cost, but all have the same goal: to recover more meaningful detail from the same or fewer sequencing reads.
Applications from viruses to tumors
These targeted methods are already reshaping several areas of biology and medicine. In infections, they can finally capture viral or bacterial RNAs that lack the poly(A) tails standard protocols expect, revealing which host cells they inhabit and how they alter host gene activity. In cancer, targeted single-cell sequencing can pinpoint which cell types carry specific mutations or fusion genes and link those to altered gene programs, helping explain why some cells become resistant to therapy. Other methods focus on alternative splicing, uncovering which cell types use which isoforms, or on rare cell populations and subtle markers that would otherwise stay below the detection threshold. In pooled CRISPR screens, improved capture of guide RNAs lets scientists link each genetic perturbation to its precise cellular response.
Choosing the right tool and what comes next
Because there is now a crowded toolbox of targeted approaches, the authors propose a decision tree to help researchers pick a method. Key questions include whether full transcriptome profiling is needed, how many genes or regions must be targeted, how far those regions sit from RNA ends, and how many cells can be processed. Looking ahead, they argue that the biggest gains will come from improving the very first capture steps, expanding clever probe-based strategies, and combining targeting with emerging long-read and direct RNA sequencing platforms. Until it becomes practical to read every RNA in every cell from end to end, targeted single-cell RNA sequencing will remain essential for seeing the parts of the cellular message that matter most for biology and disease.
Citation: Moro, G., Brunner, E. & Basler, K. A practical guide to targeted single-cell RNA sequencing technologies. Commun Biol 9, 250 (2026). https://doi.org/10.1038/s42003-026-09675-y
Keywords: single-cell RNA sequencing, targeted sequencing, transcriptomics, cancer mutations, spatial transcriptomics