Clear Sky Science · en
Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression
Why scarring lungs matters to everyone
When the lungs develop stubborn scars, breathing becomes a daily struggle. This condition, known as pulmonary fibrosis, affects millions and currently has no cure—only drugs that slow the damage. In this study, researchers uncover a previously hidden molecular “brake” inside immune cells called macrophages that helps keep lung scarring in check. Understanding this brake could open the door to new treatments not only for lung fibrosis, but potentially for other diseases where harmful scarring and runaway inflammation go hand in hand.
A tale of shape-shifting immune cells
Macrophages are frontline immune cells that patrol tissues, clear debris, and help repair damage. But they are also shape-shifters: in some situations they become pro-inflammatory fighters, while in others they turn into wound-healers that can drive scar formation. A particular scar-promoting type, called profibrotic macrophages (Fib-Mac), is strongly linked to lung fibrosis. These cells produce molecules that activate fibroblasts, which then lay down excessive collagen and other matrix components, slowly stiffening the lung. The authors wanted to know how the genetic “settings” inside macrophages decide whether they become these dangerous Fib-Mac cells or remain in more balanced, protective states.

An epigenetic brake hidden in the genome
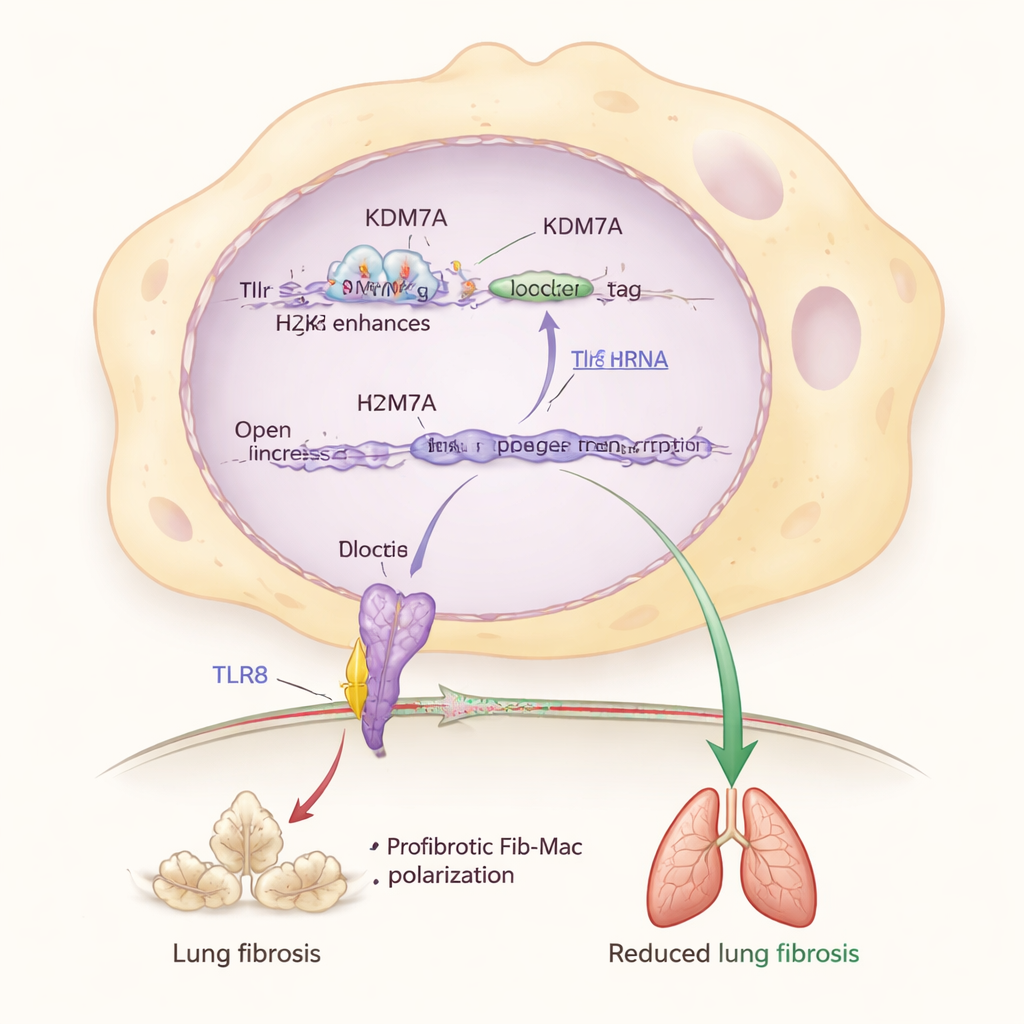
The team began by scanning hundreds of known epigenetic regulators—proteins that fine-tune how tightly DNA is packaged and which genes are turned on or off. Using RNA sequencing in both human and mouse macrophages, they found that an enzyme called KDM7A was strongly switched on when macrophages were pushed toward a fibrotic, wound-healing state. KDM7A is a “histone demethylase”: it removes certain chemical tags from histone proteins around which DNA is wrapped. That pattern suggested KDM7A might act as a built-in feedback brake, activated precisely when macrophages start drifting toward a scar-promoting identity.
To test this, the researchers used mice that lack the Kdm7a gene and triggered lung injury with the chemotherapy drug bleomycin, a standard model of pulmonary fibrosis. Early after injury, lung tissue looked similar in normal and Kdm7a-deficient animals. But by three weeks, mice missing Kdm7a showed much more extensive scarring, collapse of the tiny air sacs, and higher “Ashcroft scores” that quantify fibrosis. Genes involved in collagen production and other fibrosis-related pathways were more active in these knockout mice, confirming that loss of Kdm7a makes lungs more vulnerable to long-lasting, damaging scar formation.
How KDM7A steers macrophages away from a scar-promoting fate
Using single-cell RNA sequencing, the authors zoomed in on individual lung cells from injured mice. They discovered that in the absence of Kdm7a, a particular macrophage subset in the lung’s supporting tissue expanded dramatically and took on a strong Fib-Mac signature, expressing genes such as Arg1, Spp1, and Trem2. Further experiments in cultured macrophages showed that removing Kdm7a boosted Fib-Mac marker genes and rewired cell metabolism toward pathways that support collagen production and sustained activation. In other words, KDM7A normally restrains both the genetic and metabolic programs that drive macrophages into a fibrosis-promoting state.
Digging deeper, the researchers identified a key partner in this brake system: a sensor protein called TLR8, which detects bits of RNA inside immune cells. They found that KDM7A helps keep the Tlr8 gene switched on by removing a repressive chemical mark (H3K27me2) from an enhancer region near Tlr8. When Kdm7a was disabled, this mark accumulated, Tlr8 levels dropped, and Fib-Mac features intensified. Directly reducing Tlr8 in macrophages also pushed them toward a fibrotic identity, while activating or overproducing TLR8 pulled them back, even when Kdm7a was missing. This places the KDM7A–TLR8 pathway at the center of a molecular circuit that protects lungs from excessive scarring.
From aging lungs to human disease
To connect these findings to people, the team examined lung tissue from patients with fibrotic lung disease. Compared with non-diseased control tissue, fibrotic lungs contained many more macrophages bearing Fib-Mac markers, but these same cells showed markedly reduced KDM7A and TLR8 levels. Re-analysis of existing single-cell datasets from patients with idiopathic pulmonary fibrosis confirmed this pattern: as Fib-Mac signatures rose, KDM7A expression fell. The researchers also mined a large mouse atlas and found that Kdm7a and Tlr8 expression in macrophages declined with age in males, mirroring the higher risk of pulmonary fibrosis in older men. This suggests that age- and sex-related weakening of the KDM7A–TLR8 brake may help explain who is most vulnerable to severe lung scarring.
What this means for future treatments
In simple terms, this work shows that our immune system carries an internal safety mechanism that prevents helpful repair cells from becoming overzealous and turning into drivers of permanent scars. KDM7A, working through TLR8, keeps macrophages from locking into a profibrotic mode and thereby helps maintain flexible, functional lung tissue after injury. When this system falters—through genetic loss, aging, or other factors—macrophages are more likely to become “scar amplifiers,” worsening fibrosis. By revealing this epigenetic brake, the study points toward new therapeutic strategies: drugs that boost KDM7A activity, mimic its effects, or carefully stimulate TLR8 could someday complement existing antifibrotic therapies and offer better protection against progressive, life-limiting lung scarring.
Citation: Funagura, N., Koga, T., Etoh, K. et al. Histone demethylase KDM7A negatively regulates fibrotic macrophage polarization and lung fibrosis progression. Commun Biol 9, 309 (2026). https://doi.org/10.1038/s42003-026-09610-1
Keywords: pulmonary fibrosis, macrophages, epigenetics, KDM7A, TLR8