Clear Sky Science · en
Enhancing kcat prediction through residue-aware attention mechanism and pre-trained representations
Why faster enzyme predictions matter
Enzymes are the tiny workhorses that keep cells – and entire industries – running. They speed up chemical reactions that power our metabolism, make medicines, and drive greener manufacturing. A key number that describes how fast an enzyme works is called the turnover number, or kcat. Measuring kcat in the lab is slow and expensive, so scientists are turning to artificial intelligence to predict it from sequence and reaction information. This paper introduces PMAK, a new AI model that not only predicts kcat more accurately than earlier tools, but also helps pinpoint which parts of an enzyme are most important for its activity.
From hard lab work to smart predictions
Traditionally, determining kcat means carefully measuring how quickly an enzyme converts its substrate into product under tightly controlled conditions, such as fixed temperature and pH. Doing this for thousands of enzymes is impractical, which limits how well we can model entire metabolic networks or design new biocatalysts. Earlier computer methods tried to fill this gap, but many relied on hand‑crafted features or only a simplified view of an enzyme and a single substrate. They often worked well only when new enzymes were very similar to those already seen in training data, and they struggled with truly new enzymes, new reactions, or engineered mutants.
Teaching computers the “language” of enzymes and reactions
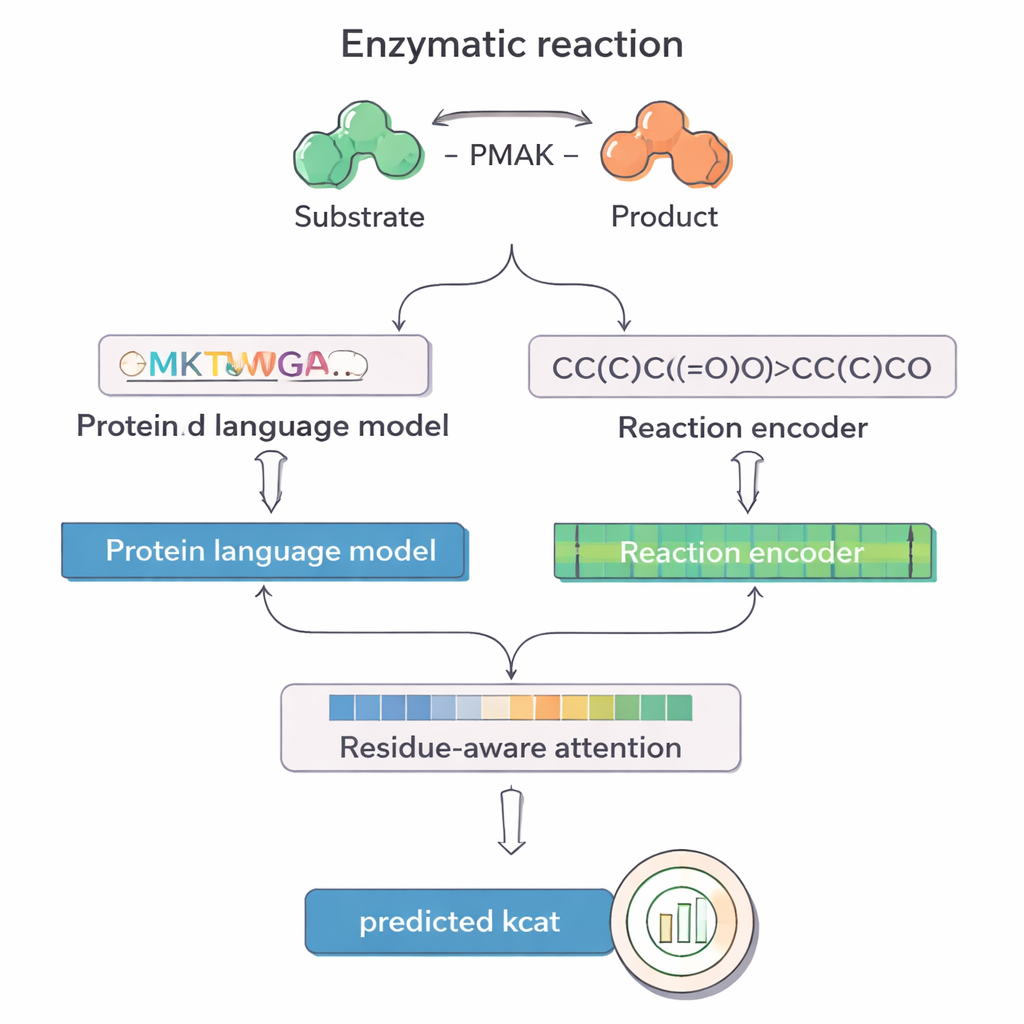
PMAK takes advantage of recent advances in “language models” originally developed for text, but retrained on vast collections of protein sequences and chemical reactions. One model, called ProT5, turns an enzyme’s amino acid sequence into a rich numerical representation that captures patterns learned from millions of proteins. Another model, RXNFP, does the same for entire reactions written as SMILES strings, which encode all reactants and products. PMAK feeds these two learned representations into a neural network that aligns their dimensions and allows the model to consider both the enzyme and the full reaction context together, rather than treating them separately.
Highlighting the most important building blocks
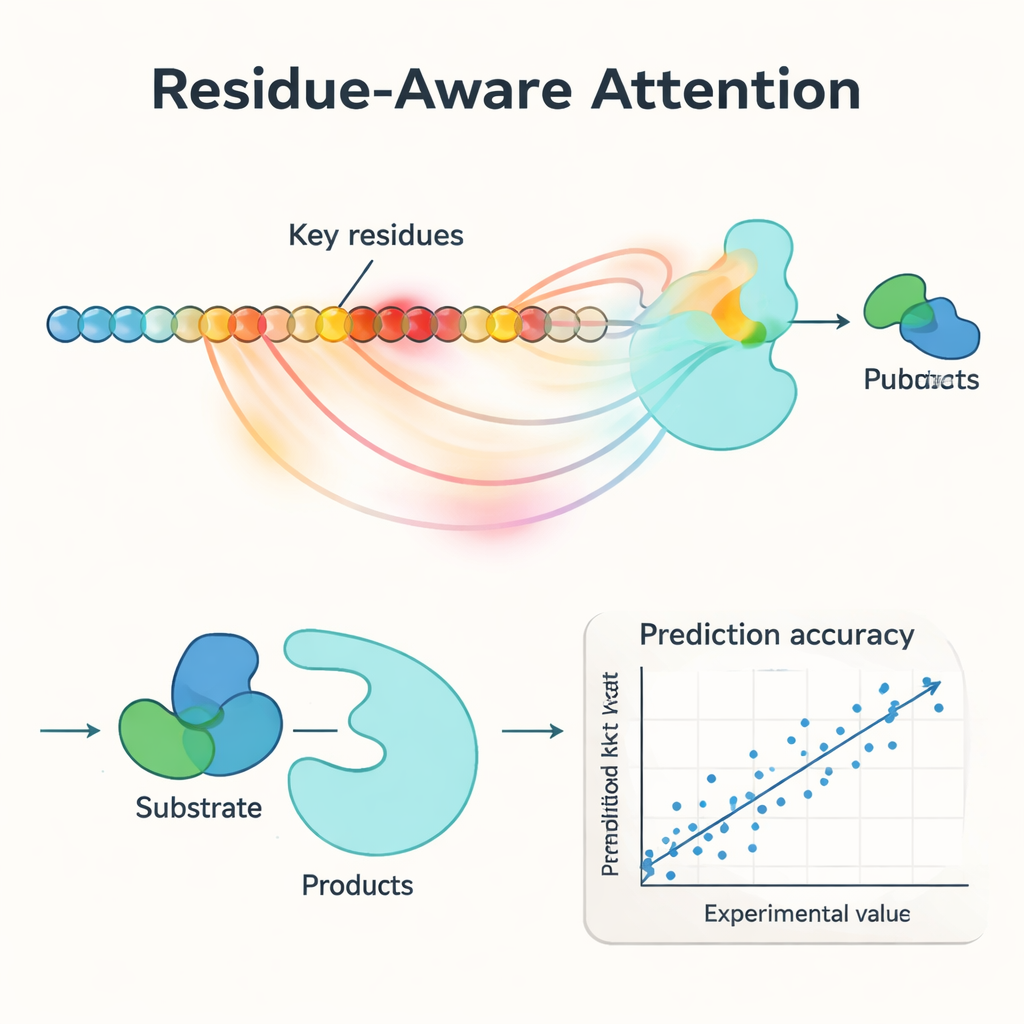
A central innovation in PMAK is a “residue‑aware attention” mechanism. Instead of treating every amino acid in an enzyme as equally important, the model learns to assign higher weights to specific residues that matter most for the reaction at hand. These attention scores act like a spotlight on the sequence: when researchers compared them with known active and binding sites from protein structures, they found that PMAK consistently highlighted functional residues far more often than chance. The model also performed well even when active sites were defined more broadly to include neighboring residues in 3D space, suggesting that it captures subtle structural and chemical cues relevant to catalysis.
Performing well on new enzymes, new reactions, and mutants
The authors rigorously tested PMAK on a curated dataset of more than 4,000 kcat values covering nearly 3,000 enzymes and 2,800 reactions. Under “warm‑start” conditions – where similar enzymes and reactions appear in both training and test sets – PMAK matched or surpassed the best existing models. More impressively, in “cold‑start” tests where either the enzyme or the reaction in the test set had never been seen before, PMAK outperformed a range of leading methods. It remained useful even for enzymes with very low sequence similarity to the training data and for reactions that looked quite different from those it had learned from. PMAK also improved predictions in realistic applications, such as estimating how cells allocate their limited protein resources and forecasting the effects of mutations in enzyme engineering datasets.
What this means for biology and biotechnology
For non‑specialists, PMAK can be viewed as a smart assistant that learns from massive protein and reaction “libraries” to guess how fast any given enzyme will work in a particular reaction – and to explain which amino acids drive that behavior. By combining stronger accuracy with residue‑level insight, this approach can help researchers design better enzymes, build more reliable metabolic models, and explore how mutations affect function without running every experiment in the lab. As similar models expand to other kinetic traits, they may become key tools for designing cleaner industrial processes, optimizing microbes for sustainable production, and deepening our understanding of how life’s molecular machines achieve their remarkable speed.
Citation: Cai, Y., Ge, F., Zhang, C. et al. Enhancing kcat prediction through residue-aware attention mechanism and pre-trained representations. Commun Biol 9, 273 (2026). https://doi.org/10.1038/s42003-026-09551-9
Keywords: enzyme kinetics, deep learning, kcat prediction, protein engineering, metabolic modeling