Clear Sky Science · en
AXL–SHC1 signaling axis mediates adaptive resistance to HER2-targeted tyrosine kinase inhibitors in HER2-aberrant lung and gastric cancers
Why some cancers outsmart new drugs
Targeted cancer drugs have transformed treatment for many patients by homing in on the specific molecules that drive tumor growth. Yet even with these precision medicines, complete and lasting remissions are uncommon. This study asks a pressing question for people with lung and stomach cancers driven by a gene called HER2: why do tumors that initially shrink on HER2-blocking pills almost always leave behind a stubborn core of cells that later fuel relapse—and how might doctors shut down that escape route from the very start?
A closer look at HER2-driven tumors
HER2 is a signaling hub that helps cells grow and survive. When it is altered or overproduced, it can turn normal cells in the breast, stomach, or lung into cancer. Several modern drugs called tyrosine kinase inhibitors (TKIs) are designed to switch off HER2 inside cancer cells. These drugs, including mobocertinib and others, can shrink tumors and delay progression. However, in lung and gastric cancers their benefits are often temporary. A small fraction of tumor cells manages to ride out the initial drug assault in a drug-tolerant state, later evolving into fully resistant tumors. Understanding what keeps these survivor cells alive is essential for designing smarter first-line treatments.
A backup lifeline called AXL
The researchers screened HER2-altered lung and gastric cancer cell lines to see which other signaling switches were helping cells withstand HER2-targeted TKIs. They identified a receptor called AXL as a key player. When cells were exposed to HER2-blocking drugs, AXL became activated and stayed switched on, even as main HER2-driven signals were dampened. This activation kept an important survival pathway, known as AKT–mTOR, running. Silencing AXL with genetic tools or blocking it with experimental drugs made the cancer cells dramatically more sensitive to multiple HER2-targeted TKIs, leading to lower cell growth and more cell death in laboratory dishes.
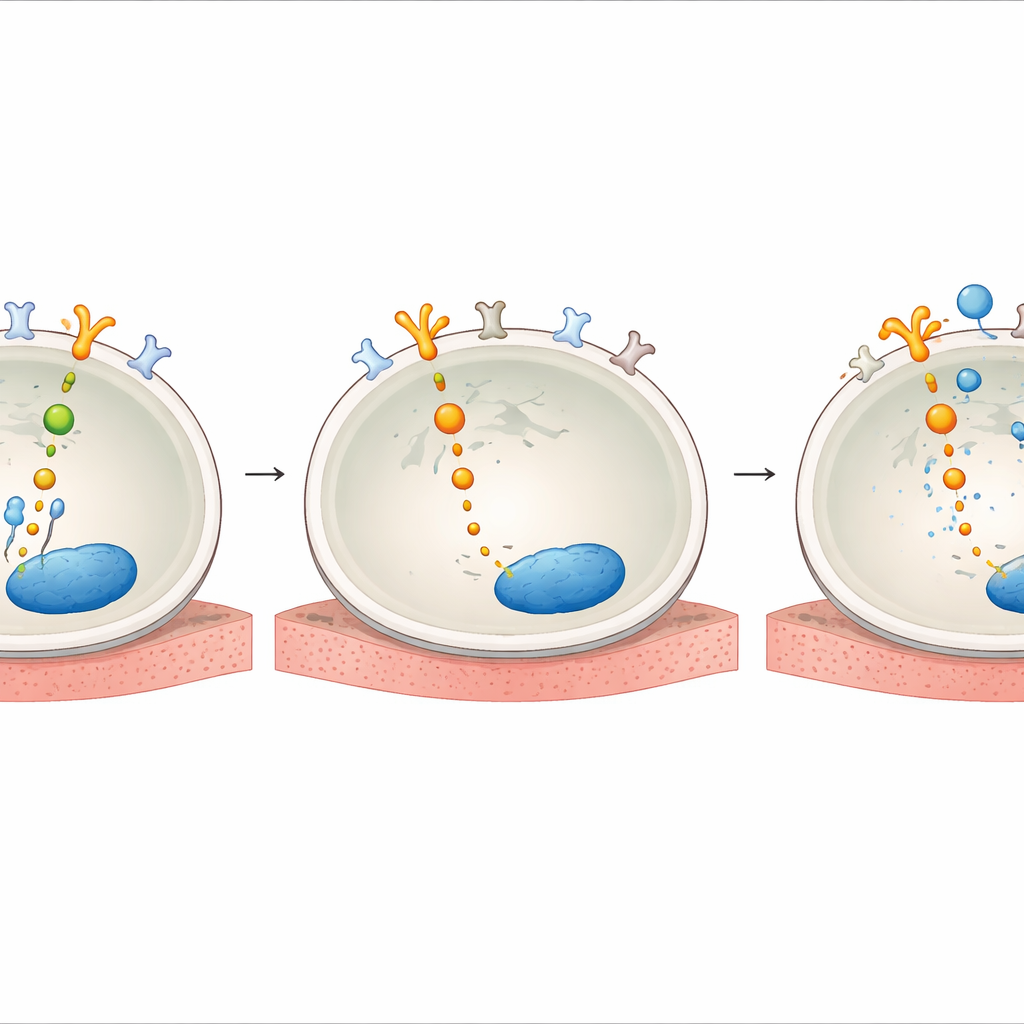
How helper proteins wire the escape route
The team then probed how AXL becomes so influential under drug pressure. They found that treatment with HER2 TKIs increased levels of GAS6, the natural partner molecule that turns on AXL at the cell surface. They also showed that, after drug exposure, AXL physically linked up with HER2 and its relatives EGFR and HER3, effectively tapping into the same survival circuitry that HER2 normally controls. Inside the cell, adapter proteins called SHC1 and SHCBP1 acted as wiring hubs. When HER2 was blocked, SHC1 detached from SHCBP1 and instead bound to AXL, while SHCBP1 moved into the nucleus, where it helped push the cell cycle forward. Knocking down SHC1 or SHCBP1 weakened AKT signaling and reduced cell survival, revealing an AXL–SHC1–SHCBP1 axis that sustains growth when HER2 is inhibited.
Stopping drug-tolerant cells before they take hold
To mimic what happens in patients, the scientists allowed cancer cells to grow for days in the presence of mobocertinib, selecting a small population that tolerated the drug. These drug-tolerant cells grew slowly but were clearly less sensitive to the HER2 inhibitor than the original cells. Their survival still depended heavily on AXL: adding an AXL blocker sharply reduced their growth and dampened AKT activity. In mouse models carrying HER2-altered, AXL-positive lung tumors, combining mobocertinib with an AXL inhibitor shrank tumors more, reduced dividing cells, and increased signs of programmed cell death compared with the HER2 drug alone—without added toxicity. Tumors engineered to overproduce AXL were notably less responsive to mobocertinib by itself, but again responded when the AXL inhibitor was added.
What this could mean for future treatment
Importantly, tissue samples from patients with HER2-altered lung and gastric cancers revealed that about a quarter had high AXL levels, and most had at least some AXL present. This suggests that a sizable group of patients might benefit from a treatment strategy that pairs a HER2-targeted TKI with an AXL inhibitor from day one, rather than waiting for resistance to appear. In plain terms, the study shows that many HER2-driven tumors keep a backup growth switch—AXL—on standby. When HER2 is blocked, AXL takes over and keeps cancer cells alive. Turning off both switches at once causes far more cancer cells to die and slows or prevents the rise of drug-tolerant survivors. If confirmed in clinical trials, this dual-target approach could lead to longer-lasting control of HER2-altered lung and stomach cancers.
Citation: Ishida, M., Yamada, T., Katayama, Y. et al. AXL–SHC1 signaling axis mediates adaptive resistance to HER2-targeted tyrosine kinase inhibitors in HER2-aberrant lung and gastric cancers. npj Precis. Onc. 10, 142 (2026). https://doi.org/10.1038/s41698-026-01385-2
Keywords: HER2-targeted therapy, AXL inhibitor, drug resistance, lung cancer, gastric cancer