Clear Sky Science · en
Midostaurin response in AML is shaped by a progenitor-like cell state selectively targeted by SMAC mimetics
Why some leukemia drugs stop working
For many people with a type of blood cancer called acute myeloid leukemia (AML), new targeted drugs have brought hope—but not everyone benefits, and responses often fade. This study asks a simple but crucial question: why do some leukemia cells ignore a widely used drug, midostaurin, and can we find a smart combination that forces these stubborn cells to die?
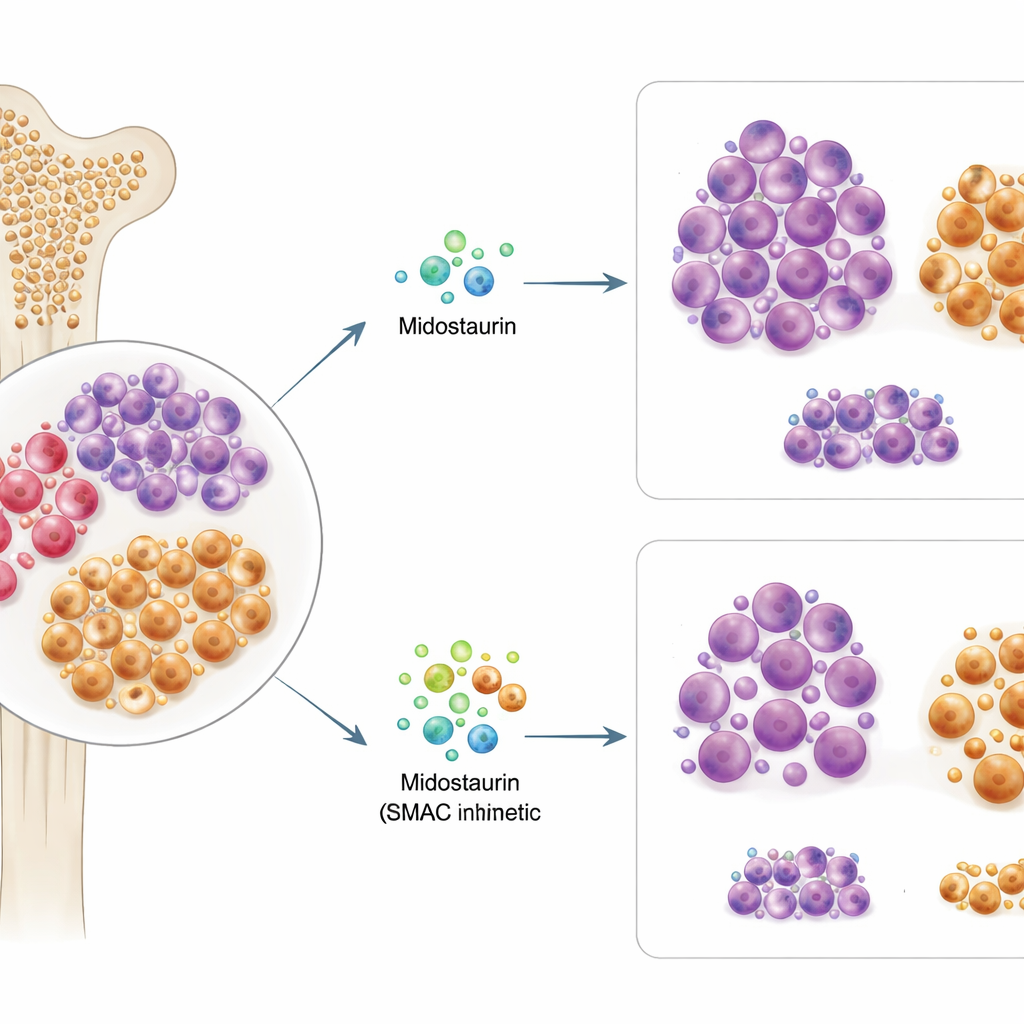
Looking beyond the main mutation
About one in three AML patients carries a change in a gene called FLT3, which drives leukemia growth and is the reason midostaurin is prescribed. The researchers tested bone marrow and blood samples from 63 patients with FLT3‑mutated AML, exposing the cells to midostaurin and more than 500 other cancer drugs in the lab. They found that how sensitive a patient’s cells were to midostaurin ex vivo closely matched how that patient later responded in the clinic. Surprisingly, the exact type of FLT3 mutation or how abundant it was did not reliably predict midostaurin success, suggesting that genetics alone cannot explain who benefits.
A hidden pool of stubborn "seed" cells
Diving deeper, the team compared the overall protein and gene activity patterns between cells that were midostaurin‑sensitive and those that were not. Non‑responding samples were enriched for features of immature, stem‑like progenitor cells—cells closer to the root of blood formation and thought to act as “seeds” that can restart the leukemia. In contrast, responding samples looked more like partially matured immune and myeloid cells. Using advanced single‑cell methods, the scientists identified a specific population of leukemia cells marked by surface proteins CD38 and CD45RA that behaved like these progenitor‑like seeds. These cells showed unusual organization of their outer membrane, hinting that key signaling molecules were being arranged in ways that favored survival.
Survival wiring: a switch of signaling pathways
Midostaurin is designed to block FLT3 signaling, which normally feeds a chain of signals that includes a molecule called STAT5 and can drive cell growth. When the team examined signaling in cell lines and patient samples after midostaurin treatment, they saw two distinct patterns. In midostaurin‑sensitive cells, STAT5 activity dropped quickly, consistent with effective FLT3 shutdown. In resistant cells, however, another pathway dominated: PI3K/AKT, a classic survival route that helps cells resist cell death. These resistant cells maintained or even increased AKT activity after treatment, and they showed higher levels of proteins that block apoptosis (programmed cell death). In other words, the wiring inside these progenitor‑like cells appeared rerouted to favor survival even when FLT3 was inhibited.
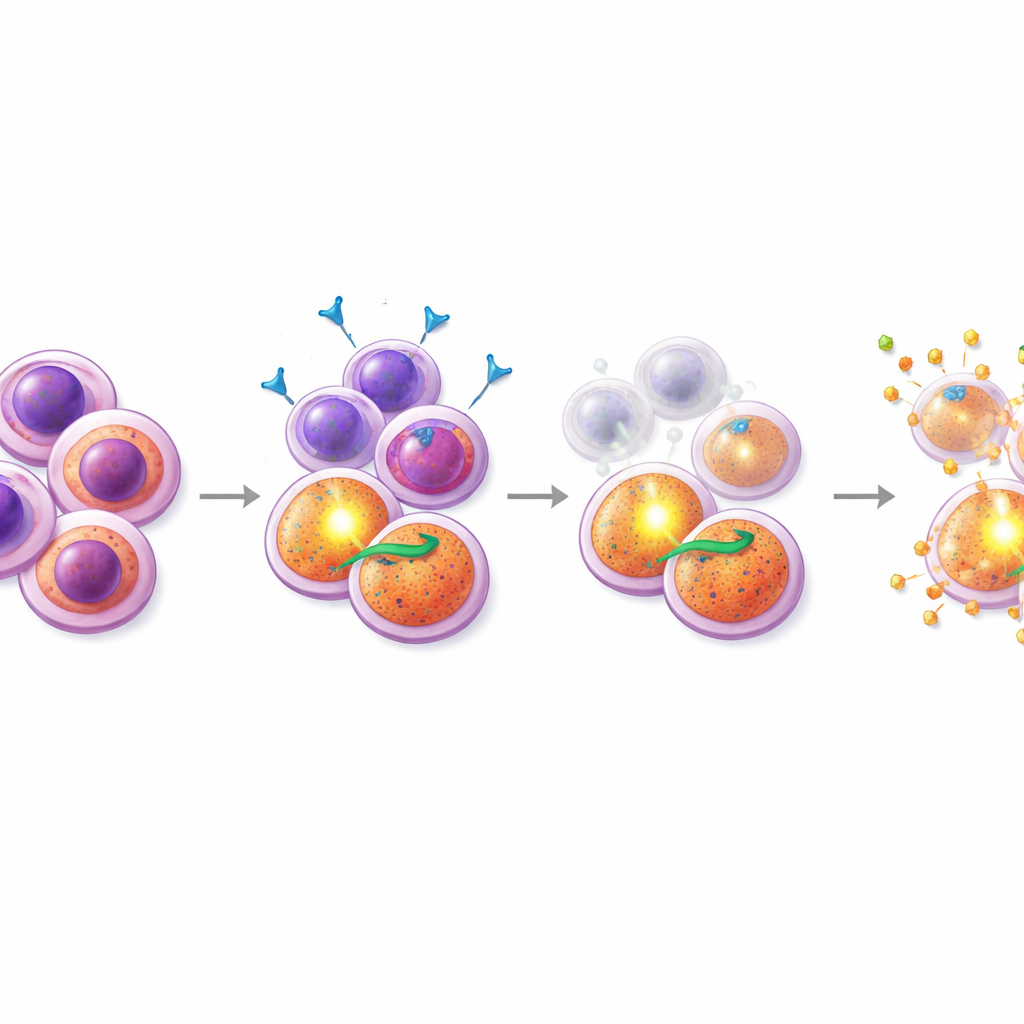
Finding a partner drug that hits the weak spot
Armed with this insight, the researchers screened combinations of midostaurin with hundreds of other compounds, focusing on drugs that influence cell death. One standout group was SMAC mimetics, drugs that disable the “inhibitor of apoptosis” proteins that PI3K/AKT‑driven cells rely on. In resistant patient samples and a resistant FLT3‑mutant cell line, adding SMAC mimetics such as birinapant to midostaurin produced strong synergy: together, the drugs killed far more cells than either alone. Crucially, detailed flow‑cytometry experiments showed that the midostaurin–SMAC mimetic combination selectively depleted the CD38+CD45RA+ progenitor‑like population and reduced its characteristic surface marker levels, suggesting that this therapy specifically targets the hard‑to‑kill seeds. By contrast, combinations with the approved BCL‑2 inhibitor venetoclax were more effective against a different, CD34‑high subset and did not show the same focused effect on the resistant cells.
What this means for patients
This work suggests that resistance to midostaurin is not just about the FLT3 mutation itself, but also about the “state” of the leukemia cells—their maturity level, membrane organization, and preferred survival pathways. A progenitor‑like CD38+CD45RA+ subset appears to be a key reservoir of resistance, shifting its signaling from the usual STAT5 route toward a survival‑friendly PI3K/AKT program. By pairing midostaurin with SMAC mimetics, the researchers were able to re‑sensitize these cells and drive them into cell death in the lab. While larger clinical studies are still needed, the findings point toward a future in which doctors might use functional testing and cell‑state profiling, not just DNA sequencing, to choose FLT3‑targeted combinations that cut off both the bulk leukemia and its most resilient seeds.
Citation: Struyf, N., Gezelius, H., Lundmark, A. et al. Midostaurin response in AML is shaped by a progenitor-like cell state selectively targeted by SMAC mimetics. npj Precis. Onc. 10, 117 (2026). https://doi.org/10.1038/s41698-026-01363-8
Keywords: acute myeloid leukemia, FLT3 inhibitors, drug resistance, leukemic stem cells, SMAC mimetics