Clear Sky Science · en
Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries
Why cleaning up sequencing data matters
Modern biology often relies on reading out millions of tiny RNA fragments to understand how cells make proteins. But these powerful measurements, especially a method called ribosome profiling (Ribo‑Seq), can be cluttered with irrelevant RNA pieces that waste sequencing power and money. This study describes a simple, data‑driven way to design specialized molecular "blockers" that selectively remove those unwanted fragments, nearly doubling the useful information researchers get from the same experiment.
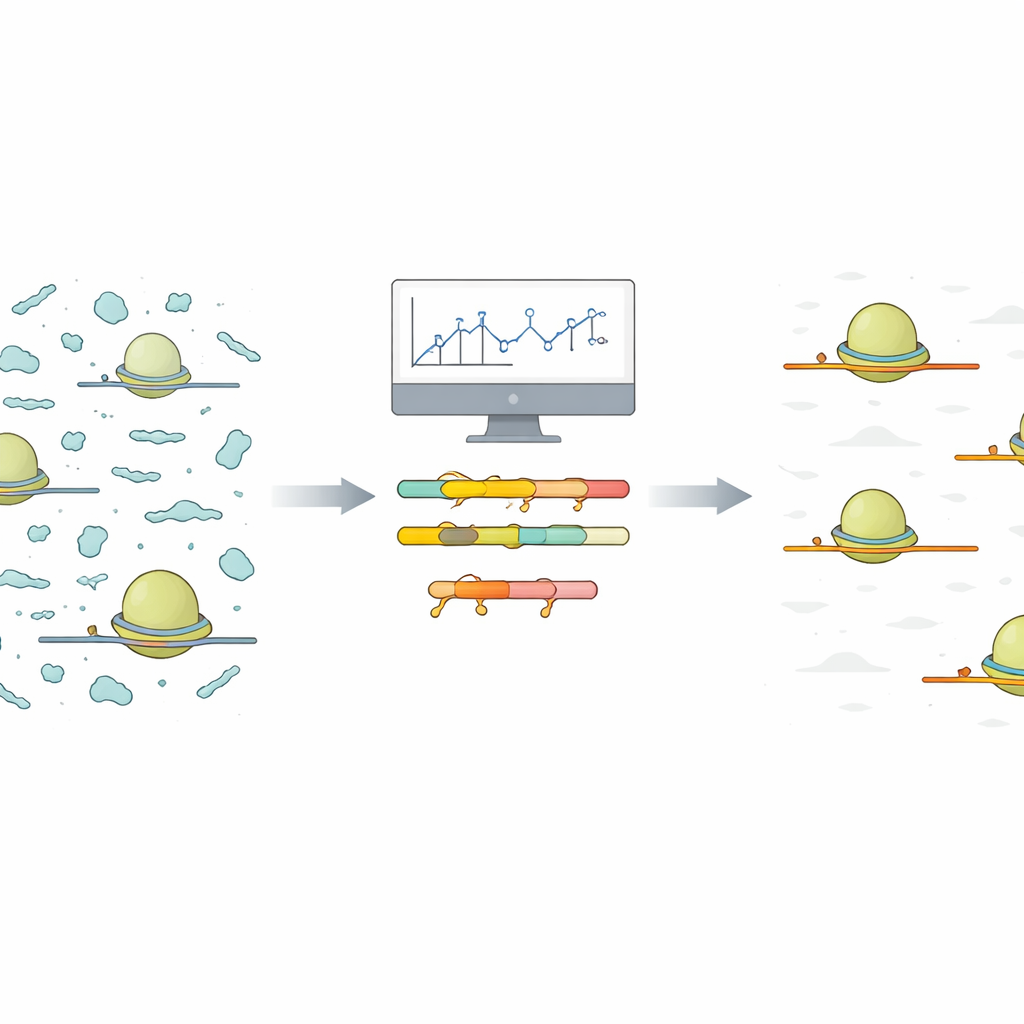
The problem of noisy ribosome snapshots
Ribo‑Seq captures a moment‑by‑moment snapshot of which messages in a cell are being actively translated into proteins. To do this, scientists isolate ribosomes together with the short stretches of messenger RNA (mRNA) they protect. Everything else is chewed up, and the protected snippets are sequenced and mapped back to the genome. In practice, however, many other small pieces of non‑coding RNA sneak through this process. Because these contaminant fragments are abundant and highly variable, they soak up a large fraction of the sequencing reads, leaving fewer reads for the true protein‑coding signals that researchers care about.
Why existing clean‑up tricks fall short
Standard strategies try to remove abundant ribosomal and other non‑coding RNAs with pre‑designed capture probes or enzymes. These methods work well when the target RNAs are intact and predictable, but Ribo‑Seq purposely chops RNA into many differently sized fragments. That fragmentation scrambles the target sites for fixed probe sets, making depletion much less efficient. On top of that, the exact mixture of contaminants depends on the species being studied, growth conditions, and even which nuclease enzyme is used. Existing clean‑up workflows also tend to involve multiple incubation and purification steps, which are time‑consuming and can cause sample loss or bias.
Custom blockers designed from real data
The authors propose a streamlined approach that starts with a small, low‑cost trial sequencing run under the same conditions planned for the full experiment. They provide an R script that takes the aligned reads from this pilot run and automatically groups similar contaminant fragments based on sequence. For each group, the script reports the shortest common sequence that appears across the fragments. These short, shared stretches are ideal target sites for specialized molecules called locked nucleic acid (LNA) oligonucleotides. LNAs are short strands with a chemical modification that makes them bind very tightly to matching RNA. The script also generates intuitive heatmaps and summary plots, helping users see which contaminants dominate and how many LNA targets would be needed for substantial clean‑up.
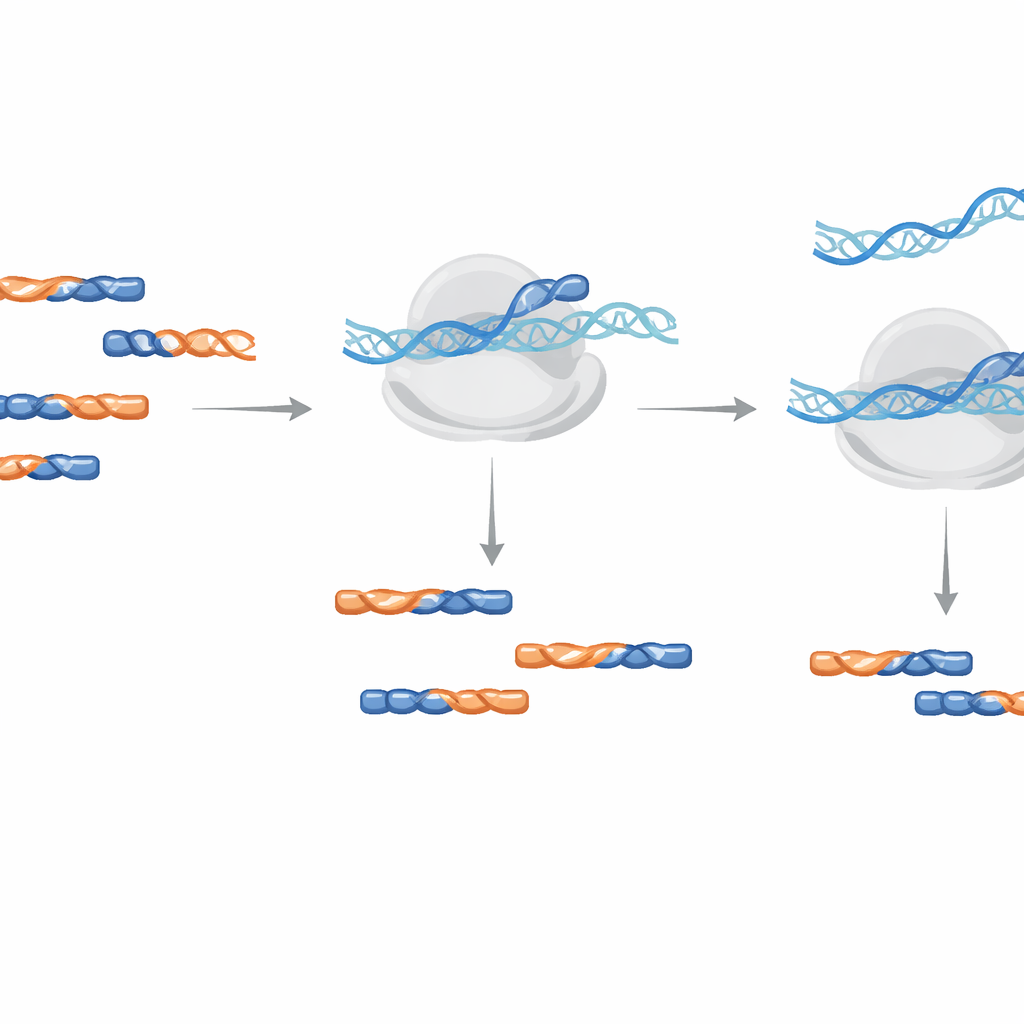
A one‑step clean‑up during amplification
Rather than physically pulling contaminants out of the sample, the method uses LNA oligonucleotides as blockers during the DNA amplification step that builds the sequencing library. The authors tested adding these blockers either during the initial reverse transcription step or during later PCR amplification. They found that adding LNAs during amplification was more efficient and required lower concentrations, reducing a test contaminant by over a thousand‑fold while working regardless of strand orientation. Practical design tips include alternating standard DNA and LNA building blocks, using a minimum length of 14 units for the plant Arabidopsis, and modifying the tail end so the blocker itself cannot be accidentally extended.
More useful reads without distorting the signal
To demonstrate real‑world performance, the team designed five LNA blockers targeting the most common contaminant groups seen across typical growth conditions in Arabidopsis plants. When they added this mix during library amplification, the proportion of identified contaminants dropped by more than 30%, and the number of useful protein‑coding reads nearly doubled. Crucially, when they compared gene‑level read counts between libraries with and without LNA treatment, the values agreed almost perfectly, indicating that the blockers removed junk fragments without distorting the biological signal from genuine mRNA footprints.
What this means for future experiments
This work shows that a short pilot experiment, coupled with an easy‑to‑use analysis script and a small set of tailored LNA blockers, can turn cluttered Ribo‑Seq libraries into much cleaner, more informative datasets in a single pipetting step. Researchers gain more meaningful reads per run, saving costs and simplifying experimental design, while preserving accurate measurements of how genes are translated. The authors also provide ready‑made contaminant profiles and blocker designs for common plant conditions, and suggest that similar resources could be built for many organisms, making high‑quality ribosome profiling more accessible across the research community.
Citation: Ricciardi, D.A., Peter, F.E. & Böhmer, M. Data-driven design of LNA-blockers for efficient contaminant removal in Ribo-Seq libraries. Sci Rep 16, 8565 (2026). https://doi.org/10.1038/s41598-026-43117-3
Keywords: ribosome profiling, RNA contaminants, locked nucleic acids, sequencing library cleanup, translation regulation