Clear Sky Science · en
Discovery of hydroxytriazole as a potential glyoxalase-I inhibitor utilizing computer-aided drug design techniques
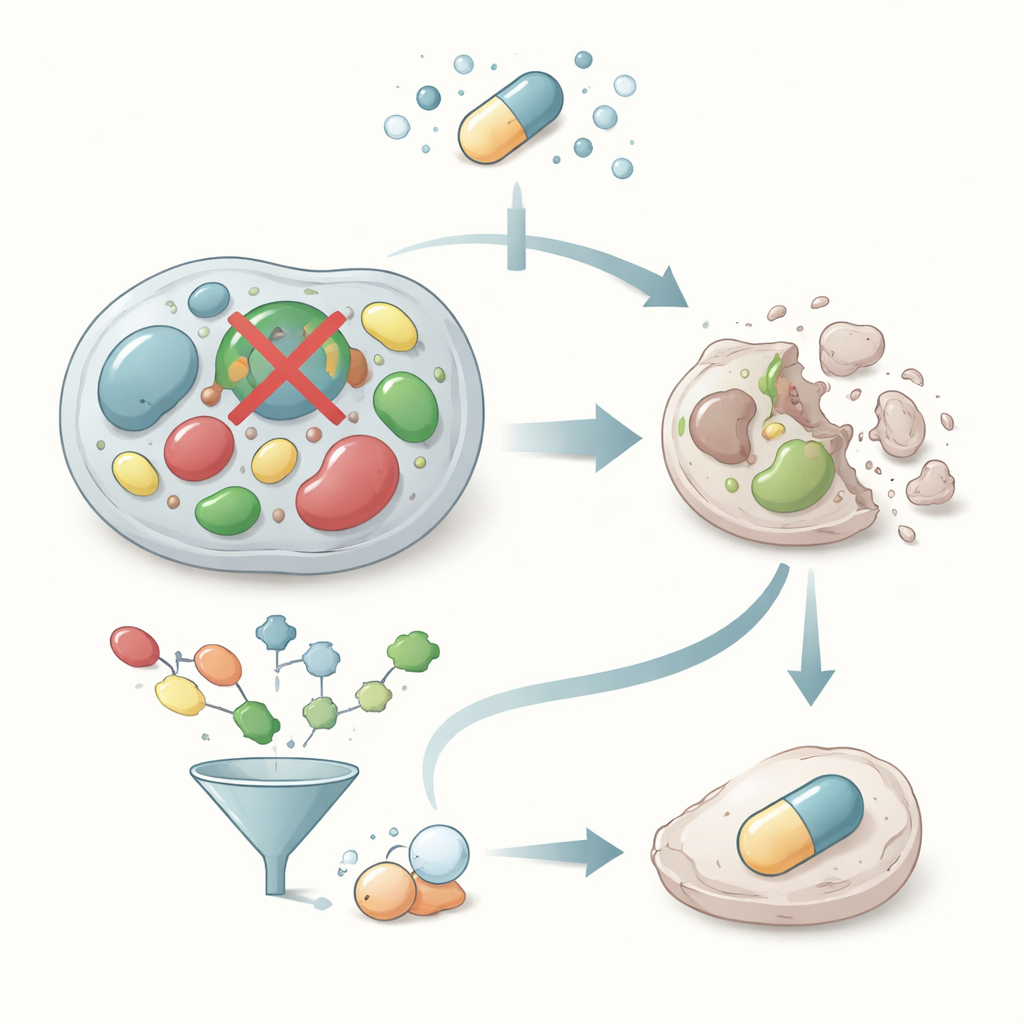
Why stopping a tiny cell cleaner could fight cancer
Cancer cells often grow so fast that they drown in their own waste. One of their survival tricks is a built‑in cleanup crew that detoxifies harmful by‑products of sugar burning. This study explores how to switch off one key member of that crew, an enzyme called glyoxalase‑I, using computers to sift through tens of thousands of molecules and experiments to test the best candidates. The goal is to discover new drug “starting points” that could one day help doctors selectively poison cancer cells from the inside.
A hidden waste system inside our cells
Every cell constantly breaks down sugar to make energy, and this process produces a reactive waste chemical called methylglyoxal. In normal amounts, our bodies convert methylglyoxal into harmless lactic acid through the glyoxalase system, a two‑step pathway that relies on the helper molecule glutathione. Glyoxalase‑I is the first and most crucial step in this chain. Cancer cells, which burn sugar at a frantic pace, depend heavily on glyoxalase‑I to keep methylglyoxal from reaching toxic levels. If this enzyme is blocked, methylglyoxal builds up and can push damaged cells toward programmed death. That makes glyoxalase‑I an appealing target for anticancer drugs that strike at a basic weakness of tumor metabolism.
Searching chemical space with silicon and statistics
Rather than testing random substances in the lab, the researchers used computer‑aided drug design to search a large commercial collection of more than 50,000 small molecules. Specialized software first cleaned and standardized each molecule, then predicted its 3D shape and behavior at body‑like pH. A fast virtual screening step scored how well each candidate might fit into glyoxalase‑I’s active site. The team then applied simple rules about size, solubility, and other drug‑likeness properties to discard molecules unlikely to work in the body. A more detailed docking program examined how the most promising molecules could orient inside the enzyme, especially how they might reach and grip the zinc metal atom that sits at the heart of glyoxalase‑I’s chemistry.
A new way to grab the enzyme’s metal core
Earlier efforts to block glyoxalase‑I focused on well‑known chemical groups, such as carboxylic acids and hydroxamic acids, that are good at binding metals but often suffer from poor stability or unwanted side effects. The present study instead uncovered a different type of “metal‑grabbing” unit: a hydroxytriazole ring. Among sixteen top‑ranked molecules chosen for purchase and lab testing, one bearing this ring—coded SPB07393SC—emerged as the standout. In virtual docking, its hydroxytriazole group reached down to the zinc atom, while its two aromatic rings tucked into nearby oily pockets of the enzyme. Computer simulations of the complex over tens of nanoseconds suggested that the molecule remained snugly bound, with stable distances, compact protein shape, and a persistent network of hydrogen bonds.
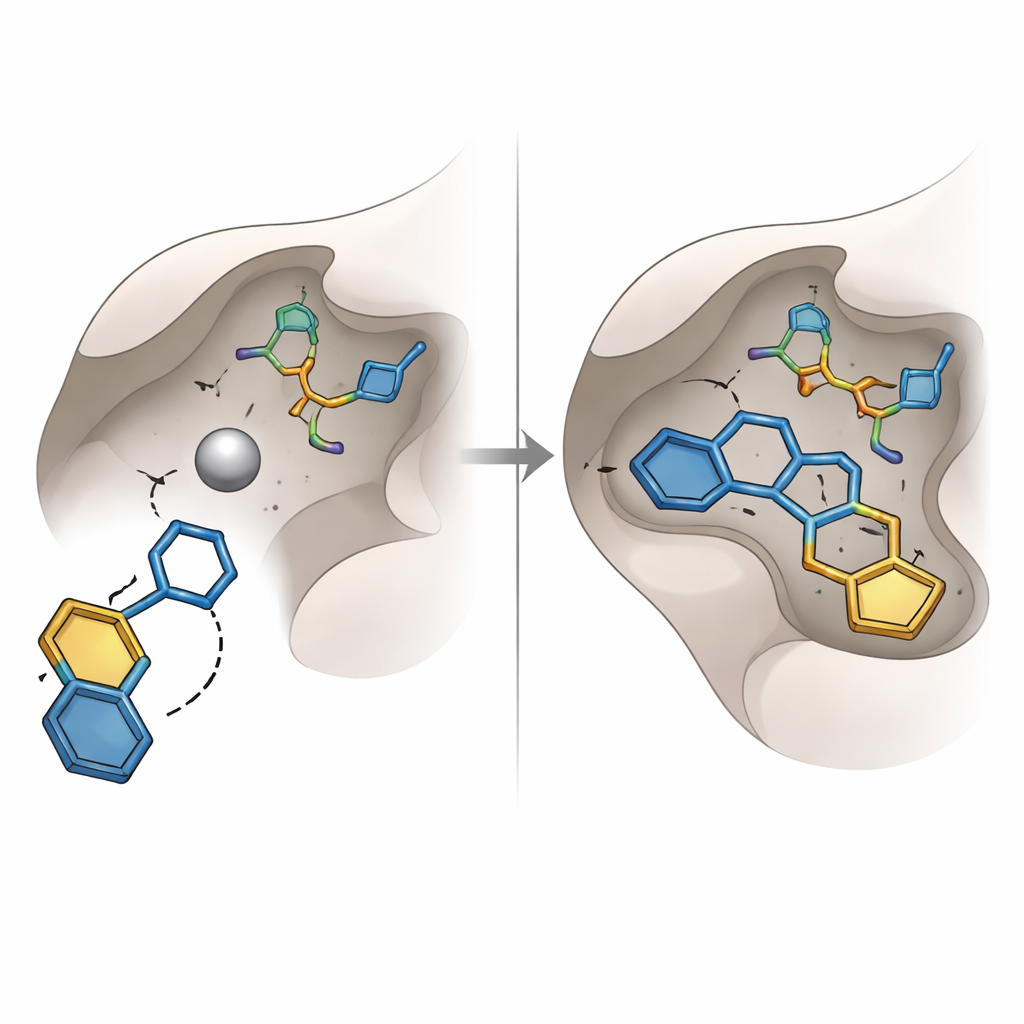
Putting the predictions to the test
To see whether the computer models translated into real‑world effects, the team measured how well the selected molecules slowed the activity of purified human glyoxalase‑I in a plate‑based assay. Fifteen of the sixteen candidates showed only weak or negligible inhibition under the tested conditions, highlighting the pitfalls of relying solely on static docking scores. In contrast, SPB07393SC inhibited the enzyme strongly, with a mid‑micromolar potency that makes it a solid early “hit” rather than a finished drug. Additional software tools predicted that this molecule should have acceptable solubility, good absorption, the ability to reach the brain if needed, and a low chance of causing certain genetic or liver‑related toxicities, though these safety predictions still require experimental confirmation.
What this means for future cancer medicines
The work introduces hydroxytriazole as a fresh way to anchor drug candidates to the zinc atom at the core of glyoxalase‑I, expanding the menu of chemical tricks available to drug designers. While SPB07393SC itself is only a starting point, its combination of enzyme blocking power, predicted drug‑like behavior, and stable binding in motion‑based simulations marks it as a promising scaffold for further fine‑tuning. More broadly, the study shows both the strengths and the limits of computer‑guided screening: it can quickly narrow vast chemical libraries to a few realistic contenders, but careful lab experiments are still essential to reveal which molecules truly disable the enzyme that cancer cells rely on to manage their toxic waste.
Citation: Al-Qazzan, M., Al-Balas, Q., Alnajjar, B. et al. Discovery of hydroxytriazole as a potential glyoxalase-I inhibitor utilizing computer-aided drug design techniques. Sci Rep 16, 9945 (2026). https://doi.org/10.1038/s41598-026-40497-4
Keywords: glyoxalase I, cancer metabolism, computer-aided drug design, zinc-binding inhibitors, molecular docking