Clear Sky Science · en
DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning
Why tiny protein pieces matter for our health
Peptides—short stretches of proteins—have become rising stars in modern medicine. They can act as precise messengers in the body and are increasingly used as drugs and disease markers. Yet figuring out which peptides are truly linked to disease usually depends on having clear examples of both “disease” and “non‑disease” peptides, something biology rarely provides. This study introduces a new way to spot potentially harmful peptides using only the ones we already know are involved in disease, offering a faster and less biased route to discovering future diagnostics and treatments.
The challenge of finding the “non-disease” group
Traditional computer models learn by comparing two sides: positive examples that are known to be disease-related and negative examples that are believed to be harmless. In peptide research, this second group is a problem. Many peptides simply have not been tested, so labeling them as “non-disease” can be misleading and introduce bias. Previous studies on anti-cancer or anti-inflammatory peptides achieved impressive accuracy, but often relied on hand-built or guessed negative datasets. As a result, their models may struggle with rare signals or new kinds of disease peptides that do not resemble the training data.
Learning from what we know, instead of what we guess
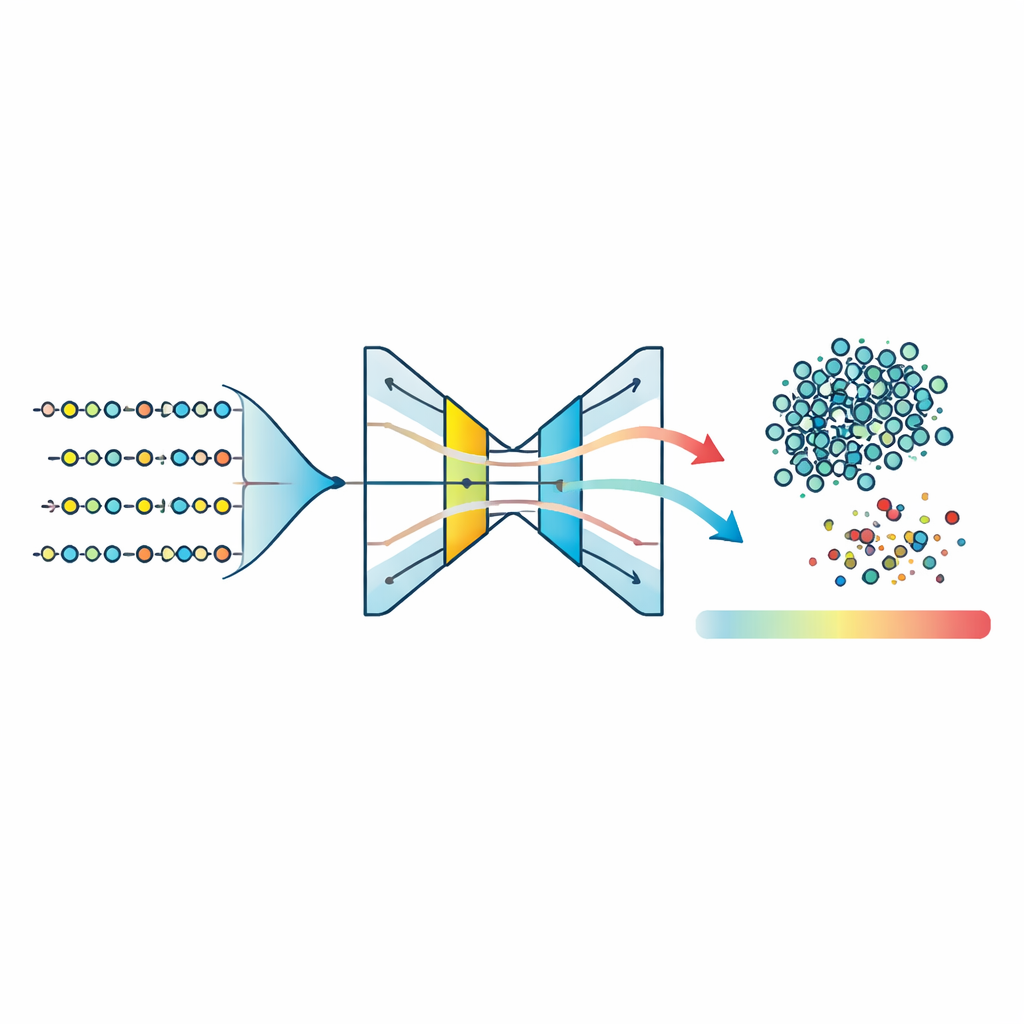
The authors take a different path: instead of forcing a two-sided problem, they treat disease-associated peptides as one coherent group and ask, “What does this group look like in detail?” They collect over 760,000 mutated human peptides from a specialized cancer-related database and describe each peptide using a rich set of features. These include how often each amino acid appears, how pairs of amino acids are arranged, basic physical and chemical traits like volume and water-loving behavior, and short recurring sequence patterns known as motifs. A technique called principal component analysis then compresses this high-dimensional description into a more manageable form while preserving the main sources of variation.
Spotting unusual peptides with one-class models
With this compressed feature space in hand, the team trains three “one-class” models—algorithms designed to learn the shape of a single group and flag anything that does not fit. They test One-Class Support Vector Machines, Isolation Forests, and a type of neural network called an autoencoder. The autoencoder learns to squeeze each peptide’s features down to a narrow internal representation and then reconstruct them; peptides that belong to the learned disease pattern are rebuilt accurately, while unusual ones incur a higher reconstruction error. Comparing normalized anomaly scores across all methods shows that the autoencoder produces the tightest cluster of typical peptides and the clearest separation between inliers and outliers. By setting a threshold on reconstruction error around the 95th percentile, the model classifies the majority of peptides as likely disease-associated while consistently flagging a small fraction as atypical.
Turning complex scores into a single, meaningful number
To make the results easier to interpret biologically, the authors introduce the Disease Peptide Anomaly Score (DPAS). This score blends two ingredients: how unusual a peptide looks to the autoencoder (its normalized reconstruction error) and how strongly its features contribute to predictions, as measured by a popular explanation method called SHAP. In practice, motifs and specific physical–chemical traits emerge as especially informative. DPAS combines these signals so that peptides that are both structurally odd and supported by biologically meaningful features receive higher ranks. The top-scoring peptides are then examined with a motif‑search tool that links them to known functional signatures such as phosphorylation sites, metal-binding regions, and other regulatory patterns commonly involved in signaling and enzyme control.
What this means for future diagnostics and drugs
In everyday terms, this work offers a smarter filter for finding suspect peptides without pretending we know which ones are definitely harmless. By learning only from confirmed disease-related examples and then ranking new candidates with DPAS, researchers can prioritize a short, biologically plausible list of peptides for laboratory testing. Many of the highest-ranked candidates contain well-known functional motifs, reinforcing the idea that they may play roles in disease processes. While the method still depends on assumptions and lacks experimentally proven “safe” peptides for full validation, it provides a more realistic and transparent foundation for peptide biomarker discovery and could be adapted to other biological data types where reliable negative examples are scarce.
Citation: Khalid, Z., Khalid, R. & Sezerman, O.U. DPAS: disease-associated peptide anomaly score for identifying pathogenic peptides via one-class learning. Sci Rep 16, 9170 (2026). https://doi.org/10.1038/s41598-026-40099-0
Keywords: disease-associated peptides, anomaly detection, autoencoder, biomarker discovery, one-class learning