Clear Sky Science · en
De novo generation and in silico screening of anti-diabetic peptide candidates via a deep learning–attention framework with physicochemical feature fusion
Why smarter peptide design matters for diabetes
Diabetes affects hundreds of millions of people worldwide and current drugs do not work perfectly for everyone. Many treatments lose effectiveness over time or cause side effects. A promising new option is a class of tiny proteins called anti-diabetic peptides, which can fine-tune blood sugar with high precision. The challenge is that finding new peptide drugs in the lab is slow and expensive. This study introduces a computer-driven pipeline that can invent and sift through large numbers of potential anti-diabetic peptides, pointing researchers toward the most promising candidates to test in the real world.
From known diabetes peptides to clean starting data
The researchers began by assembling a high-quality collection of peptides that have been experimentally shown to affect blood sugar, mostly by influencing hormones such as GLP-1 or enzymes like DPP-IV. These formed the “positive” examples. They then built a matching “negative” set of peptides with no reported anti-diabetic activity, carefully chosen so that length, composition, and basic chemistry resembled the positives. To avoid fooling the model with near-duplicates, they used sequence-similarity tools to make sure that closely related peptides never appeared in both training and testing groups. This homology-aware splitting ensured that the system would be judged on its ability to recognize genuinely new patterns rather than memorizing old ones.
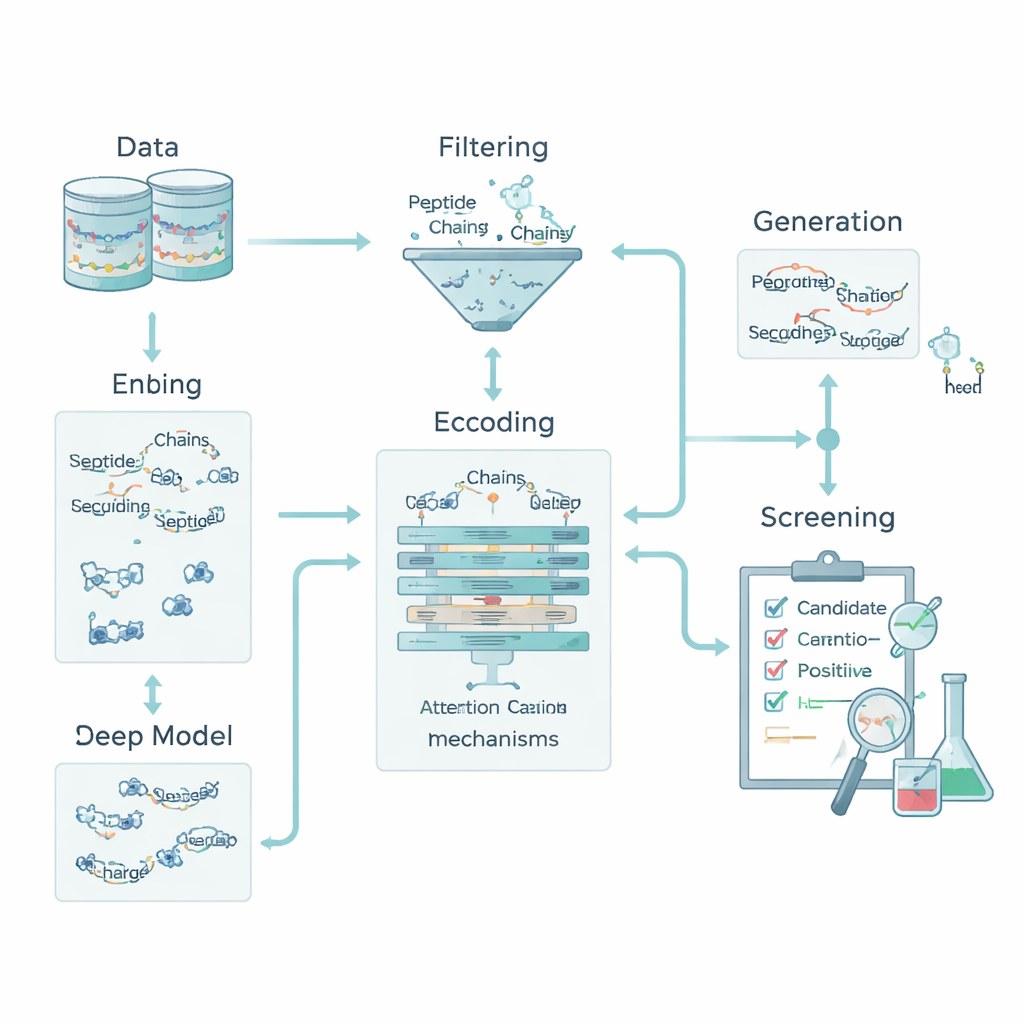
Encoding chemistry so machines can read peptides
For a computer, a peptide is just a string of letters representing amino acids. To connect those letters to biology, the team transformed each amino acid into five basic chemical traits: how water-repelling it is, its electrical charge, its tendency to form hydrogen bonds, its mass, and whether it has an aromatic ring. This turned every peptide into a small “image” that captures both order and chemistry. On top of this, they added whole-peptide descriptors such as overall charge, average hydrophobicity, and the Boman index, which relates to how strongly a peptide tends to bind to other proteins. Together, these features let the model look at both local patterns—short motifs of amino acids—and global properties that influence how a peptide behaves in the body.
A deep learning engine that explains its choices
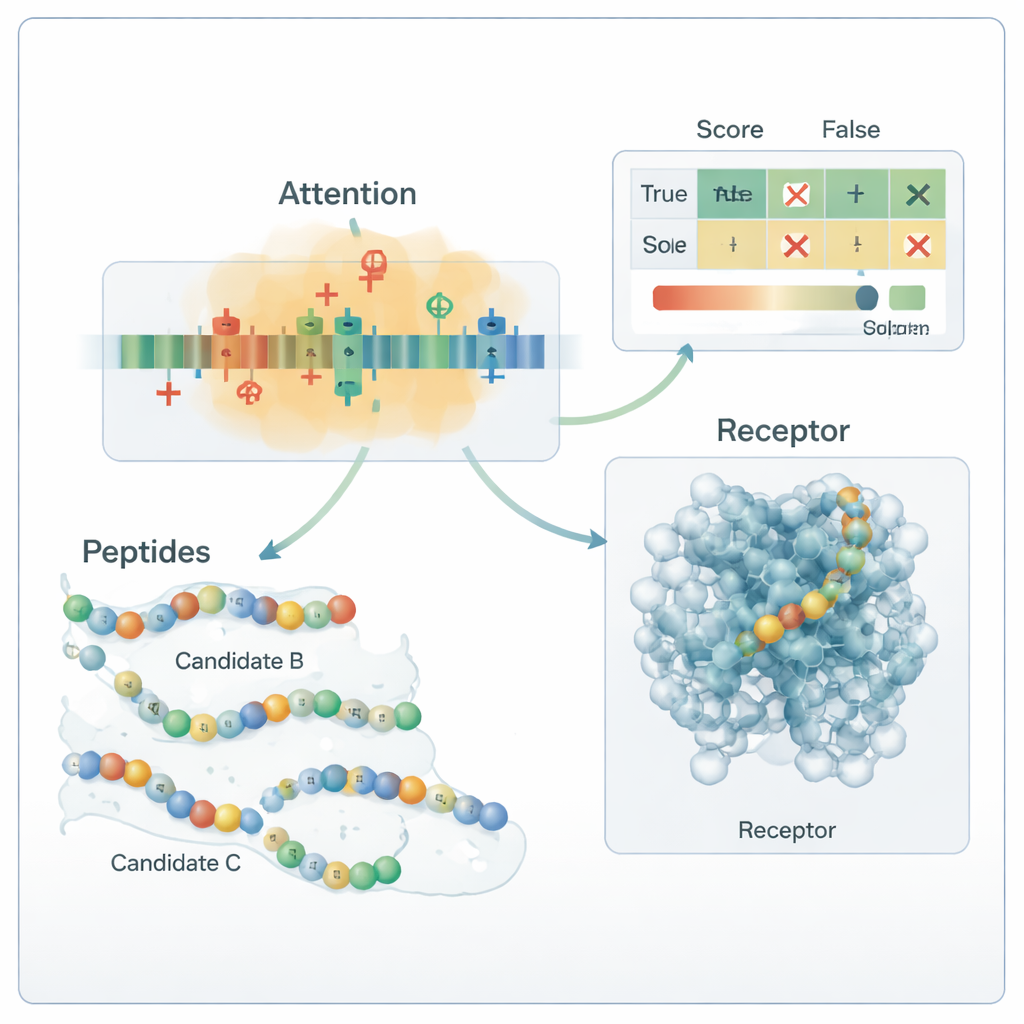
The core of the pipeline is a hybrid deep learning model. A convolutional neural network (CNN) scans along the peptide, hunting for short motifs that tend to appear in active peptides, much like filters in an image-recognition system. On top of that, an attention layer learns which positions in the sequence matter most, capturing long-range relationships between distant residues. The output of this sequence engine is fused with the global chemical descriptors and passed to several standard machine-learning classifiers—support vector machines, decision trees, k-nearest neighbors, and gradient-boosted trees. A specialized optimization method, called OptimizedTPE, automatically tunes their settings, striking a balance between accuracy and the risk of overfitting. The attention mechanism also gives residue-level “importance maps,” helping scientists see which parts of each peptide drive the model’s decisions.
Inventing new candidates while avoiding data leakage
To overcome the small number of known anti-diabetic peptides, the team added a generation stage that only feeds the training process. They used a mix of strategies—guided mutation, motif recombination, and a variational autoencoder—to propose new sequences that resemble, but do not copy, known active peptides. These candidates were then screened through strict “descriptor gates” that enforce realistic charge, size, and binding propensity, plus external tools that score similarity to known bioactive peptides. Only sequences that pass these filters and remain clearly distinct from all test peptides are kept as weakly labeled positives for training; none are ever used to evaluate the model. This approach expanded the training set while preserving a clean, unbiased test bed.
How well the system works and what it means
When challenged with an entirely independent panel of 180 experimentally studied peptides collected from recent literature, the framework correctly labeled roughly 99 out of 100 sequences, with both precision and recall near 0.99. In practical terms, that means it rarely misses a true anti-diabetic peptide and rarely calls an inactive peptide promising. Analyses of the attention maps and mutation tests showed that the model has learned chemically sensible rules: it relies heavily on positively charged and certain hydrophobic residues known to be important for binding diabetes-related targets. Molecular docking simulations further suggested that some of the newly generated peptides can make plausible contacts with the human GLP-1 receptor. While these predictions still require laboratory confirmation, the study demonstrates a reproducible, biologically grounded way to explore the vast space of possible peptide drugs and to prioritize the few that are most likely to help manage diabetes.
Citation: Asl, Z.R., Rezaee, K., Ansari, M. et al. De novo generation and in silico screening of anti-diabetic peptide candidates via a deep learning–attention framework with physicochemical feature fusion. Sci Rep 16, 6580 (2026). https://doi.org/10.1038/s41598-026-39985-4
Keywords: anti-diabetic peptides, deep learning, drug discovery, peptide design, GLP-1 receptor