Clear Sky Science · en
Ferrostatin 1 exerts multifaceted hepatic protection against alcoholic liver injury by inhibiting ferroptosis
Why this matters for people who drink
Many people know that heavy drinking can damage the liver, but the exact reasons why some livers fail while others cope for years are still being uncovered. This study looks at a newly discovered type of cell death called ferroptosis and tests a small protective molecule, Ferrostatin‑1, in mice. By showing how this compound shields the liver from alcohol‑driven damage on several fronts at once—fat buildup, iron overload, oxidative stress, and inflammation—the work points to fresh ways doctors might one day slow or prevent alcoholic liver disease rather than just urging people to stop drinking and hoping the liver recovers on its own.
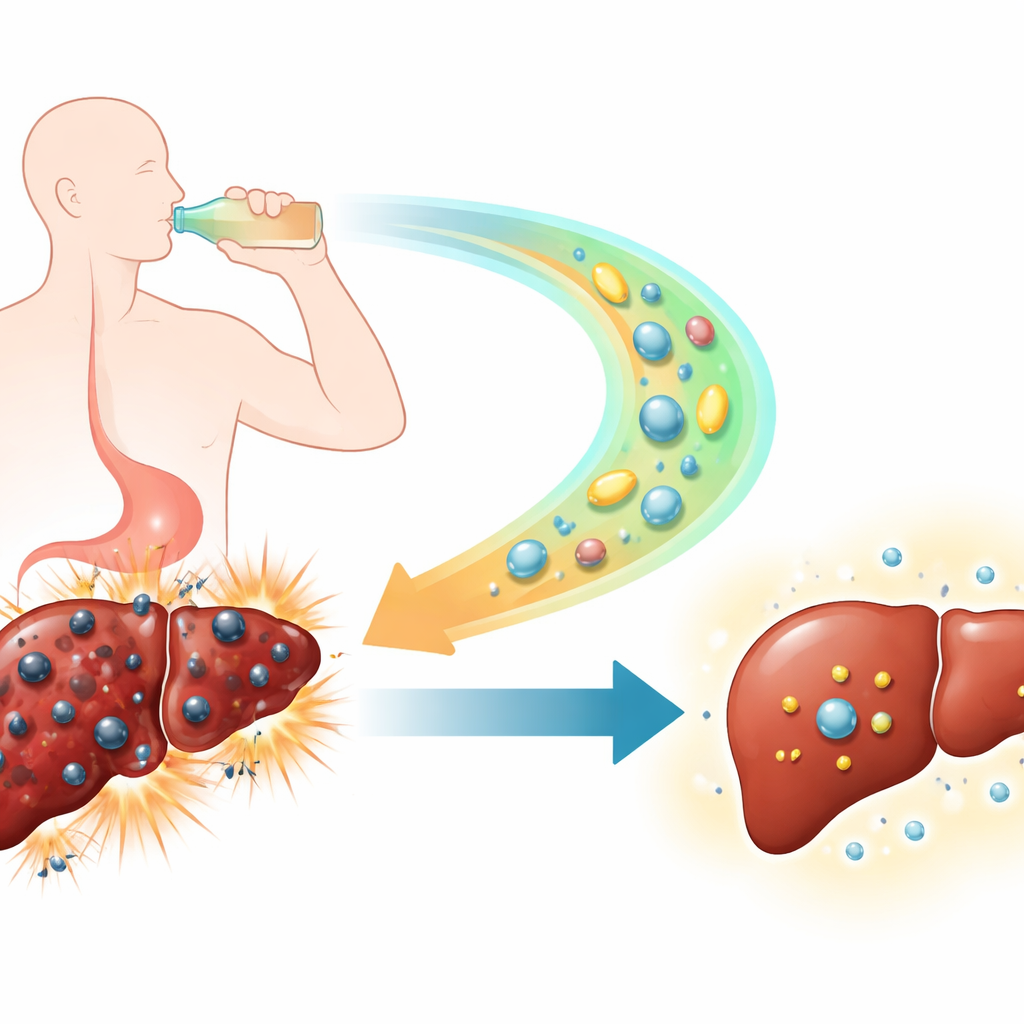
A new kind of cell death in a stressed liver
Traditional views of liver damage from alcohol have focused on familiar forms of cell death such as apoptosis and necrosis. Over the past decade, however, scientists have identified ferroptosis, a distinct form of cell death that relies on iron and the runaway oxidation of fats in cell membranes. In this study, the authors used mouse models that mimic both short binge episodes and long‑term heavy drinking in humans. They found clear signs that ferroptosis is active in the livers of alcohol‑exposed animals: iron levels and reactive oxygen species rose sharply, protective antioxidants such as glutathione dropped, and chemical by‑products of fat oxidation accumulated. Together, these changes marked liver cells that were being pushed into an iron‑driven, self‑amplifying death spiral.
A small molecule that breaks the damage cycle
To see whether blocking ferroptosis could spare the liver, the researchers treated some of the alcohol‑fed mice with Ferrostatin‑1, a compound designed to soak up the highly reactive fat radicals that drive this process. In both the short‑term and long‑term drinking models, Ferrostatin‑1 improved standard blood measures of liver function and reduced the scarring, fat droplets, and structural disruption seen under the microscope. At the molecular level, the drug restored the activity of key protective proteins (GPX4 and SLC7A11) and replenished glutathione, helping cells neutralize harmful oxidants before they shredded membrane lipids. These results suggest that Ferrostatin‑1 does more than merely dampen damage; it directly interferes with the core machinery of ferroptosis.
Untangling fat, iron, and inflammation
Alcoholic liver disease does not arise from a single flaw; rather, it reflects a tangled web of disturbed fat handling, mismanaged iron, and chronic immune activation. The study shows that ferroptosis sits at the center of this web. Mice given alcohol developed marked fat accumulation in their livers and in the bloodstream, along with excess iron both in liver tissue and in circulation. Signals that normally balance iron traffic—such as the hormone hepcidin, the iron exporter ferroportin, the iron importer transferrin receptor, and the iron‑storage protein ferritin—were all skewed in ways that trap iron in the liver. Ferrostatin‑1 largely reversed these shifts, easing iron overload and fat buildup at the same time. In parallel, it reduced the influx and activation of immune cells in the liver and lowered inflammatory messengers that promote tissue injury and fibrosis, hinting that dying ferroptotic cells are powerful triggers of inflammation.
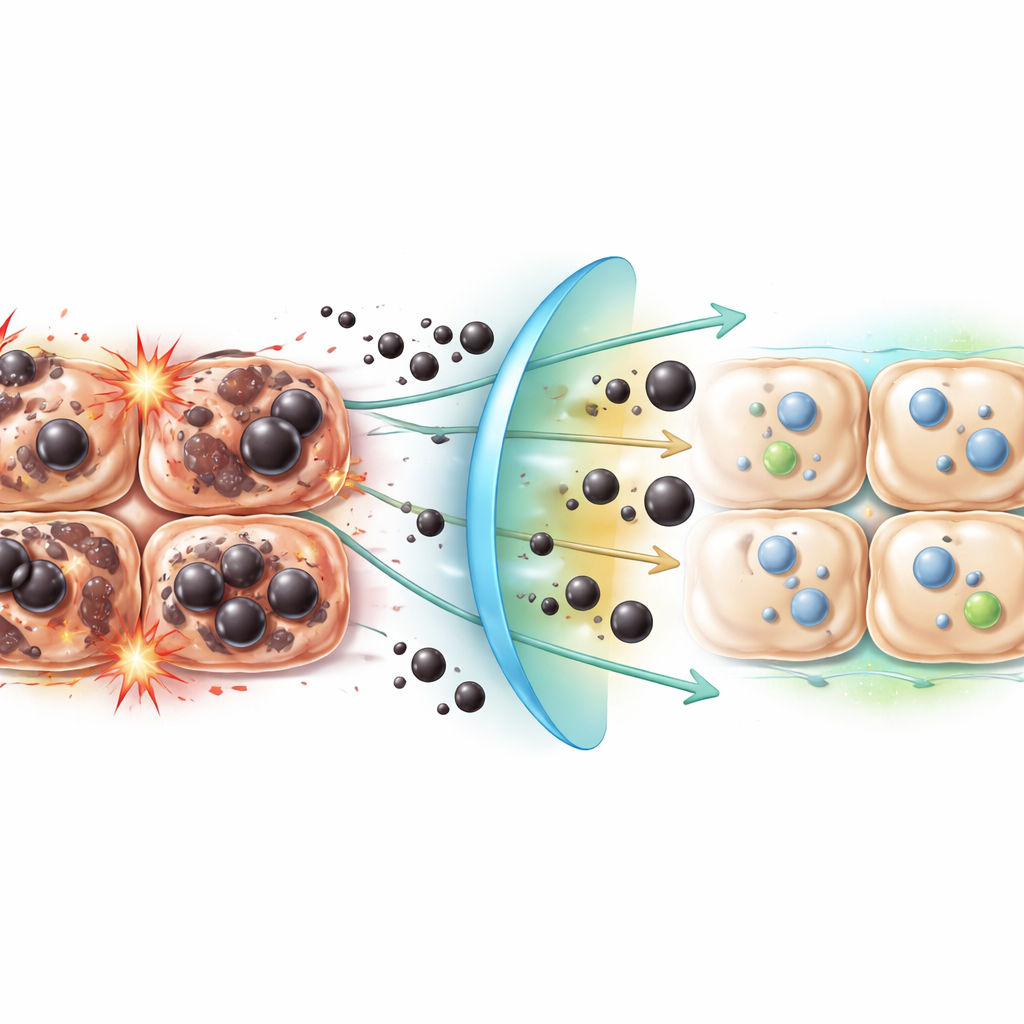
Working independently of well‑known safety switches
Our cells deploy several broad defense systems to counter stress, including a master antioxidant controller called Nrf2, the recycling process known as autophagy, and a protein complex named the NLRP3 inflammasome that helps set off inflammatory alarms. Earlier work suggested these pathways could influence ferroptosis. Surprisingly, in the alcohol‑fed mice, Ferrostatin‑1 delivered strong liver protection without noticeably changing Nrf2, its partner protein HO‑1, autophagy markers, or NLRP3 activity. This implies that the compound acts in a more direct and focused way—by intercepting the specific lipid radicals that define ferroptosis—rather than by broadly cranking up the cell’s stress responses. It also hints that ferroptosis may drive liver injury through channels that only partially overlap with classic antioxidant and inflammatory circuits.
What this could mean for future treatments
For people struggling with alcohol use, stopping drinking remains the most important step, and this study does not change that. But it does show that alcoholic liver damage is not just passive wear and tear; it is powered by an active, iron‑fueled form of cell death that can be interrupted. In mice, Ferrostatin‑1 broke a vicious cycle in which alcohol pushed liver cells toward ferroptosis, which in turn worsened iron imbalance, fat overload, and inflammation. Although this compound itself is not ready for use in humans, the findings build a strong case for developing drugs that safely target ferroptosis in the liver. Such therapies could one day complement abstinence and other medical care, helping vulnerable livers stay functional longer and reducing the progression toward cirrhosis and liver cancer.
Citation: Yu, L., Zhang, H., Wang, Y. et al. Ferrostatin 1 exerts multifaceted hepatic protection against alcoholic liver injury by inhibiting ferroptosis. Sci Rep 16, 9145 (2026). https://doi.org/10.1038/s41598-026-39849-x
Keywords: alcoholic liver disease, ferroptosis, iron overload, oxidative stress, liver inflammation