Clear Sky Science · en
Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction
Why smarter cancer drugs matter
Cancer drugs often behave like blunt tools: while they attack tumor cells, they can also hit healthy tissues and cause serious side effects. One promising way to sharpen this aim is to block specific versions of an enzyme called carbonic anhydrase, which helps tumors survive in low-oxygen environments. However, several versions of this enzyme look almost identical, making it hard to design medicines that hit the “bad” ones in tumors without disturbing the “good” one found throughout the body. This study shows how interpretable machine learning can help researchers navigate this challenge and design more selective, safer drug candidates.
The problem of hitting the wrong target
Human carbonic anhydrase (hCA) comes in many forms, or isoforms. Two of them, IX and XII, are linked to the survival of cancer cells in oxygen-starved tumors, so blocking them could slow disease and improve treatment. But isoform II is widespread in healthy tissues and has an active site that looks very similar to IX and XII. Drugs that bind all three can trigger unwanted problems such as metabolic acidosis and vision disturbances. Traditional lab and computer methods struggle because enzymes are large, complex molecules, and the number of possible drug-like compounds is astronomically high. Exhaustively testing them all, either in the lab or on a computer, is simply not feasible.
Building a clean and trustworthy data foundation
The authors tackled this by first assembling a carefully cleaned database of thousands of molecules tested against hCA II, IX, and XII from the ChEMBL repository. They standardized chemical structures, removed dubious measurements, and focused on compounds that share a common zinc-binding group typical of this class of inhibitors. Using strict cutoffs, they labeled molecules as clearly active or clearly inactive and discarded borderline cases that could confuse the models. Because there were far more inactive than active molecules, they balanced the data so that learning algorithms would not simply favor the majority class. They also used a “scaffold-based” way of splitting the data so that training and test sets contained different core molecular frameworks, giving a more realistic picture of how well the models would handle truly new compounds.
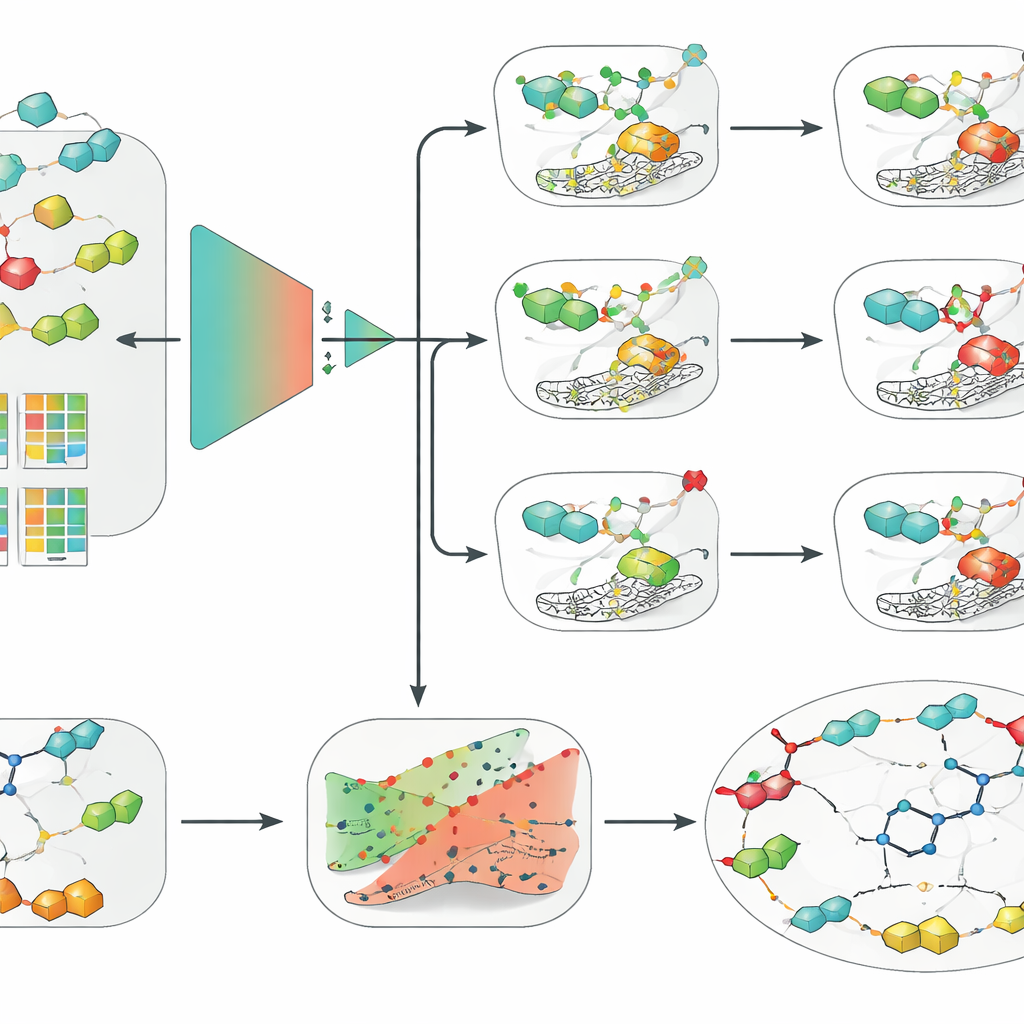
Simple models beat deep learning when data are limited
With this curated dataset, the team compared a wide range of approaches, from classic machine learning methods like logistic regression, random forests, and support vector machines (SVMs) to modern deep neural networks, including graph-based models that operate directly on molecular structures. They paired these with several ways of encoding molecules, such as traditional hand-crafted descriptors, key-based fingerprints, and learned embeddings from a chemical language model. Across all three enzyme isoforms and under the stricter scaffold-based evaluation, one combination consistently stood out: an SVM fed with extended-connectivity fingerprints, a structured way of describing local chemical environments within a molecule. Surprisingly, this comparatively simple setup outperformed more fashionable graph and deep learning models, highlighting that data quality, careful validation, and good molecular descriptors can matter more than algorithmic complexity when datasets are of modest size.
Adding reliable confidence and human-friendly explanations
The researchers then wrapped their best SVM model in two additional layers designed to make its predictions more usable in real drug discovery. First, they applied a framework called conformal prediction, which does not just output a single yes-or-no answer but instead provides a region of likely outcomes together with a guaranteed error rate. This allows scientists to tune how cautious they want the model to be and to recognize cases where the model is genuinely uncertain. Second, they used counterfactual explanations to make the model’s reasoning more intuitive. For a given molecule, they generated closely related analogs that flip the predicted outcome from active to inactive, or vice versa. Examining these pairs for the clinical candidate SLC-0111, which selectively blocks IX and XII but not II, the method independently rediscovered an important medicinal chemistry insight: small changes in the “tail” part of the molecule strongly alter which isoform it prefers to bind.
From algorithms to practical drug design tools
To make their approach accessible, the authors packaged the three SVM models, the uncertainty layer, and the counterfactual engine into a graphical tool called CAInsight. A user can provide a molecule’s text representation and, with a single click, obtain predicted activity against hCA II, IX, and XII, an estimate of how trustworthy each prediction is, and suggested structural tweaks that might increase or reduce activity. While the models focus on classifying molecules as active or inactive rather than predicting exact potency or selectivity in one step, they already reproduce known behavior for real drug candidates and distinguish subtle structural changes. The authors note that larger and more uniform datasets, plus a deeper analysis of how activity cutoffs are chosen, could further refine performance.
What this means for future cancer medicines
In plain terms, this work shows that carefully built and well-explained machine learning models can help chemists design cancer drugs that better distinguish between look-alike enzyme targets. By combining robust statistics, uncertainty estimates, and intuitive “what-if” examples, the framework not only predicts which molecules are likely to work but also suggests why. This kind of transparent artificial intelligence could speed up virtual screening, support generative design of new compounds, and reduce the trial-and-error burden in the lab, ultimately aiding the discovery of more selective and safer treatments for patients.
Citation: Ghamsary, M.S., Rayka, M. & Naghavi, S.S. Interpretable machine learning rationalizes carbonic anhydrase inhibition via conformal and counterfactual prediction. Sci Rep 16, 8419 (2026). https://doi.org/10.1038/s41598-026-39771-2
Keywords: carbonic anhydrase inhibitors, interpretable machine learning, drug selectivity, conformal prediction, counterfactual explanations